
用于治疗视网膜变性的基于CRISPR/CAS9的组合物和方法与流程

本申请要求于2016年7月5日提交的美国临时申请号62/358,337的权益,所述美国临时申请的全部内容在此通过引用并入。
背景
视网膜变性是尤其包括利伯氏先天性黑蒙(lca)、视网膜色素变性(rp)和青光眼的一组病症。lca是可遗传形式的视网膜变性,其特征在于在生命的前几个月期间严重的视网膜功能障碍和严重的视力损害。lca是罕见疾病(影响少于200,000美国人的疾病),但lca的18种亚型一起是遗传性失明的更常见原因。指定lca10的亚型是更常见的亚型,占所有lca病例的>20%。某些形式的lca顺应通过重组腺伴随病毒(aav)的治疗,所述重组腺伴随病毒(aav)经改造为递送缺陷型细胞基因的功能性拷贝。在2008年,在i期临床试验中,将补充rpe65中的突变的转基因通过aav成功地递送至lca2患者(maguiream等人nengljmed.2008;358(21):2240-2248)。注意到一些应答,但这些应答不是持久的,因为转基因表达更终丧失(schimmerj等人humgenetherclindev.2015;26(4):208-210;azvolinskya.natbiotechnol.2015;33(7):678-678)。此外,引起不同lca亚型的一些基因对于aav递送而言简直太大。因此,这些lca亚型仍然无法治愈。
adrp构成所有rp病例的大约30-40%,并且在adrp患者中,更常见的突变rp相关基因是编码视杆视色素视紫红质的基因(dryja,t.p.等人thenewenglandjournalofmedicine323,1302-1307(1990);dryja,t.p.等人nature343,364-366(1990))。目前,不存在fda批准的用于adrp患者的治疗;然而,许多方法正在开发中。这些方法中的大多数是关于“抑制和替代”主题的变化。在这种方法中,敲低负责变性的基因的表达,例如用核酶或经由rna干扰(rnai)敲低视紫红质rna的水平(shrna和sirna方法两者正在探索中),然后用“硬化的”基因替代内源等位基因的表达,所述“硬化的”基因不易被核酶或rnai试剂敲低。可能更接近临床的这一主题的变体是由genabletechnologieslimited开发的rhonova试剂。rhonova采用sirna来敲低与aav递送的cdna组合的内源性视紫红质表达(突变体和野生型两者),所述cdna编码不易被sirna敲低的经修饰的但功能性视紫红质(http://www.genable.net)。
青光眼,全世界不可逆失明的首因(levkovitch-verbinh等人iovsorg44,3388–3393(2003)),是视神经病变,其中在视神经头的筛板(laminacribosa)处的视网膜神经节细胞(rgc)轴突的进行性损伤导致轴突变性和细胞死亡(howellgr等人jcellbiol179,1523–1537(2007))。目前,**的治疗,无论是通过滴眼液、激光还是切口手术,都是降低眼内压(iop)并且减少在视神经头处的损伤。不幸的是,这在一些患者中是困难的,而在其他患者中,尽管侵袭性iop降低,该疾病仍可继续恶化。该领域长期以来需要替代治疗策略,其通过减轻对残余轴突损伤的rgc应答来补充iop降低。此外,nei已将视神经再生列为其大胆的目标(audaciousgoal),并且任何再生疗法都必须解决视神经切断的rgc存活的问题。为此,非常需要开发可能直接干扰rgc轴突变性和/或轴突损伤相关细胞死亡的活跃遗传程序的神经保护剂(adalbertr等人science(2012),doi:10.1126/science.1223899;yangj等人cell160,161–176(2015);welsbieds等人procnatacadsciusa110,4045–4050(2013);watkinsta等人procnatacadsciusa110,4039–4044(2013))。
因此,非常需要用于治疗视网膜变性如lca、adrp和青光眼的新型和改进的疗法。
发明概述
除非另有说明,否则本发明的实践通常采用细胞生物学、细胞培养、分子生物学、转基因生物学、微生物学、重组核酸(例如dna)技术、免疫学和rna干扰(rnai)的常规技术,所述常规技术在本领域的技术内。这些技术中的某些的非限制性描述在下述出版物中发现:ausubel,f.,等人(编辑),currentprotocolsinmolecularbiology、currentprotocolsinimmunology、currentprotocolsinproteinscience和currentprotocolsincellbiology,均为johnwiley&sons,n.y.,截止2008年12月的版本;sambrook,russell和sambrook,molecularcloning.alaboratorymanual,第3版,coldspringharborlaboratorypress,coldspringharbor,2001;harlow,e.和lane,d.,antibodies—alaboratorymanual,coldspringharborlaboratorypress,coldspringharbor,1988;freshney,r.i.,“cultureofanimalcells,amanualofbasictechnique”,第5版,johnwiley&sons,hoboken,n.j.,2005。关于治疗剂和人类疾病的非限制性信息可在goodmanandgilman'sthepharmacologicalbasisoftherapeutics,第11版,mcgrawhill,2005,katzung,b.(编辑)basicandclinicalpharmacology,mcgraw-hill/appleton&lange第10版(2006)或第11版(2009年7月)中找到。关于基因和遗传病症的非限制性信息在以下中找到:mckusick,v.a.:mendelianinheritanceinman.acatalogofhumangenesandgeneticdisorders.baltimore:johnshopkinsuniversitypress,1998(第12版)或更近的在线数据库:onlinemendelianinheritanceinman,omim?.mckusick-nathansinstituteofgeneticmedicine,johnshopkinsuniversity(baltimore,md.)和nationalcenterforbiotechnologyinformation,nationallibraryofmedicine(bethesda,md.),截至2010年5月1日,可获自万维网:http://www.ncbi.nlm.nih.gov/omim/和onlinemendelianinheritanceinanimals(omia),动物物种(除人和小鼠外)的基因、遗传性病症和性状数据库,可获自万维网:http://omia.angis.org.au/contact.shtml。本文提及的所有**、**申请和其他出版物(例如,科学论文、书籍、网站和数据库)通过引用整体并入。在说明书与任何引用的参考文献之间冲突的情况下,以说明书(包括其任何修改,其可以基于引用的参考文献)为准。除非另有说明,否则本文使用标准的本领域公认的术语含义。本文使用关于各种术语的标准缩写。
本文描述了用于治疗视网膜变性的方法,所述视网膜变性例如视神经病,包括利伯氏先天性黑蒙(例如,利伯氏先天性黑蒙10cep290突变(lca))、视网膜色素变性(例如视紫红质r135突变)或青光眼。该方法使用修饰的核酸酶系统,例如成簇规律间隔短回文重复(clusteredregularlyinterspacedshortpalindromicrepeats)(crispr)(例如crispr相关(cas)9(crispr-cas9、非cas9crispr系统、crispr-cpf-1系统等等),以剪切和/或修复基因组dna或rna(例如,cas13a/c2c2系统)。基于crispr系统的基因编辑可以用于失活或纠正引起视神经病变和视网膜变性的基因突变(例如,lca和视紫红质突变),从而提供了用于这些疾病组的基因治疗方法。在一些实施方案中,crispr系统用于引入突变,所述突变使引起视网膜变性(例如青光眼)的正常基因(例如dlk和/或lzk)失活。因为这些基因在损伤感知和细胞存活中起作用,所得到的效应是细胞存活。“神经保护”方法不是相互排斥的方法,因为存在同样可能导致青光眼的基因突变,并且这些将与视网膜变性相同。在一些实施方案中,青光眼的突变靶包括但不限于optn、tbk1、tmco1、pmm2、gmds、gas7、fndc3b、txnrd2、atxn2、cav1/cav2、p16ink4a、six6、abca1、afap1和cdkn2b-as。
因此,本发明的一个方面涉及用于治疗影响主体的视网膜区域的病症(例如,视网膜变性)的方法,该方法包括向主体的视网膜区域施用治疗有效量的核酸酶系统,其包含基因组靶向核酸酶和包含至少一种靶向基因组序列的指导dna。
本发明的另一个方面提供了利用组合物用于治疗视网膜变性的方法,所述组合物包含先前在wo2015/195621(通过引用整体并入本文)中描述的非天然存在的crispr系统的修饰。此类修饰使用靶向视网膜变性相关基因的某些grna,所述基因例如但不限于lca10cep290基因、视紫红质、双亮氨酸拉链激酶(dlk)、亮氨酸拉链激酶(lzk)、jnk1-3、mkk4、mkk7、atf2、jun、mef2a、sox11或puma。在一些实施方案中,组合物包含(a)包含一种或多种载体的非天然存在的核酸酶系统(例如,crispr),所述载体包含:i)与编码核酸酶系统指导rna(grna)的至少一种核苷酸序列可操作地连接的启动子(例如,双向h1启动子),其中所述grna与主体的细胞中的dna分子的靶序列杂交,并且其中所述dna分子编码在细胞中表达的一种或多种基因产物;以及ii)在细胞中可操作的调节元件,其与编码基因组靶向核酸酶(例如cas9蛋白)的核苷酸序列可操作地连接,其中组件(i)和(ii)位于系统的相同或不同载体上,其中所述grna靶向所述靶序列且与之杂交,并且核酸酶切割dna分子以改变一种或多种基因产物的表达。在一些实施方案中,将系统包装到单个腺伴随病毒(aav)颗粒内。在一些实施方案中,启动子包含:a)提供在编码grna的至少一种核苷酸序列的一个方向上的转录的控制元件;以及b)提供在编码基因组靶向核酸酶的核苷酸序列的相反方向上的转录的控制元件。
本发明的另一个方面提供了改变真核细胞中的一种或多种基因产物表达的方法,其中所述细胞包含编码一种或多种基因产物的dna分子,所述方法包括向所述细胞内引入先前在wo2015/195621(通过引用整体并入本文)中描述的修饰的非天然存在的crispr系统。此类修饰使用某些grna,其靶向视网膜变性相关基因,例如但不限于lca10cep290基因、视紫红质、双亮氨酸拉链激酶(dlk)、亮氨酸拉链激酶(lzk)、jnk1-3、mkk4、mkk7、atf2、jun、mef2a、sox11或puma。在一些实施方案中,该方法包括向细胞中引入组合物,所述组合物包含(a)包含一种或多种载体的非天然存在的核酸酶系统(例如,crispr),所述载体包含:i)与编码核酸酶系统指导rna(grna)的至少一种核苷酸序列可操作地连接的启动子(例如,双向h1启动子),其中所述grna与主体的细胞中的dna分子的靶序列杂交,并且其中所述dna分子编码在细胞中表达的一种或多种基因产物;以及ii)在细胞中可操作的调节元件,其与编码基因组靶向核酸酶(例如cas9蛋白)的核苷酸序列可操作地连接,其中组件(i)和(ii)位于系统的相同或不同载体上,其中所述grna靶向所述靶序列且与之杂交,并且核酸酶切割dna分子以改变一种或多种基因产物的表达。在一些实施方案中,将系统包装到单个腺伴随病毒(aav)颗粒内。在一些实施方案中,启动子包含:a)提供在编码grna的至少一种核苷酸序列的一个方向上的转录的控制元件;以及b)提供在编码基因组靶向核酸酶的核苷酸序列的相反方向上的转录的控制元件。
本发明的一个方面涉及用于治疗有此需要的主体中的视网膜变性的方法,该方法包括:(a)提供包含一种或多种载体的非天然存在的核酸酶系统,所述载体包含:i)与编码核酸酶系统指导rna(grna)的至少一种核苷酸序列可操作地连接的启动子,其中所述grna与主体的细胞中的dna分子的靶序列杂交,并且其中所述dna分子编码在细胞中表达的一种或多种基因产物;以及ii)在细胞中可操作的调节元件,其与编码基因组靶向核酸酶的核苷酸序列可操作地连接,
其中组件(i)和(ii)位于系统的相同或不同载体上,其中所述grna靶向所述靶序列且与之杂交,并且核酸酶切割dna分子以改变一种或多种基因产物的表达;以及(b)向主体的视网膜区域施用治疗有效量的系统。
在一些实施方案中,该系统是crispr。
在一些实施方案中,将系统包装到单个腺伴随病毒(aav)颗粒内。
在一些实施方案中,该系统使一种或多种基因产物失活。
在一些实施方案中,核酸酶系统切除至少一个基因突变。
在一些实施方案中,启动子包含双向启动子。在一些实施方案中,启动子包含与选自seqidno:739-787的核苷酸序列具有至少80%、85%、90%、95%、98%、99%或同一性的核苷酸序列。在一些实施方案中,启动子包含选自seqidno:739-787的核苷酸序列。
在一些实施方案中,双向启动子是h1(seqidno:787)。h1启动子既是polii又是poliii启动子。在一些实施方案中,启动子与h1启动子直向同源。
在一些实施方案中,直向同源h1启动子衍生自真哺乳亚纲哺乳动物。
在一些实施方案中,直向同源h1启动子衍生自大熊猫(ailuropodamelanoleuca)、普通牛(bostaurus)、普通狨(callithrixjacchus)、家犬(canisfamiliaris)、豚鼠(caviaporcellus)、绿猴(chlorocebussabaeus)、霍氏树懒(choloepushoffmanni)、九带犰狳(dasypusnovemcinctus)、奥氏更格卢鼠(dipodomysordii)、家马(equuscaballus)、普通刺猬(erinaceuseuropaeus)、家猫(feliscatus)、大猩猩(gorillagorilla)、智人(homosapiens)、十三纹地松鼠(ictidomystridecemlineatus)、非洲象(loxodontaafricana)、恒河猴(macacamulatta)、小家鼠(musmusculus)、雪貂(mustelaputoriusfuro)、小棕蝠(myotislucifugus)、白颊长臂猿(nomascusleucogenys)、北美鼠兔(ochotonaprinceps)、穴兔(oryctolaguscuniculus)、小耳大婴猴(otolemurgarnettii)、绵羊(ovisaries)、黑猩猩(pantroglodytes)、东非狒狒(papioanubis)、苏门答腊猩猩(pongoabelii)、岩石蹄兔(procaviacapensis)、大狐蝠(pteropusvampyrus)、褐家鼠(rattusnorvegicus)、野猪(susscrofa)、眼镜猴(tarsiussyrichta)、树鼩(tupaiabelangeri)、宽吻海豚(tursiopstruncatus)、羊驼(vicugnapacos)。
在一些实施方案中,直向同源h1启动子衍生自小鼠或大鼠。
在一些实施方案中,直向同源h1启动子包含与seqidno:752-786中所示的核苷酸序列具有至少80%、85%、90%、95%、98%、99%或同一性的核苷酸序列。
在一些实施方案中,直向同源h1启动子包含由seqidno:752-786组成的组中所示的核苷酸序列。
在一些实施方案中,h1启动子包含:a)提供在编码grna的至少一种核苷酸序列的一个方向上的转录的控制元件;以及b)提供在编码基因组靶向核酸酶的核苷酸序列的相反方向上的转录的控制元件。
在一些实施方案中,基因组靶向的核酸酶是cas9蛋白。
在一些实施方案中,cas9蛋白进行密码子优化用于在细胞中表达。
在一些实施方案中,启动子与至少一个、两个、三个、四个、五个、六个、七个、八个、九个或十个grna可操作地连接。
在一些实施方案中,视网膜区域是视网膜。
在一些实施方案中,细胞是视网膜感光细胞。
在一些实施方案中,细胞是视网膜神经节细胞。
在一些实施方案中,视网膜变性选自利伯氏先天性黑蒙(lca)、视网膜色素变性(rp)和青光眼。
在一些实施方案中,视网膜变性是lca1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17或18。
在一些实施方案中,视网膜变性是lca10。
在一些实施方案中,靶序列在lca10cep290基因中。
在一些实施方案中,靶序列是cep290基因中的突变。
在一些实施方案中,靶序列选自seqidno:1-109、164-356、735-738中所示的核苷酸序列,或其组合。
在一些实施方案中,靶序列包含可操作地连接的seqidno:1、2、3和4。
在一些实施方案中,载体包含seqidno:110中所示的核苷酸序列。
在一些实施方案中,视网膜变性是常染色体显性形式的视网膜色素变性(adrp)。
在一些实施方案中,一种或多种基因产物是视紫红质。
在一些实施方案中,靶序列是视紫红质基因中的突变。
在一些实施方案中,靶序列是在视紫红质基因的r135处的突变。
在一些实施方案中,在r135处的突变选自r135g、r135w、r135l。
在一些实施方案中,靶序列选自seqidno:111-126中所示的核苷酸序列、或其组合。
在一些实施方案中,grna序列选自seqidno:127-142中所示的核苷酸序列、或其组合。
在一些实施方案中,视网膜变性是青光眼。
在一些实施方案中,一种或多种基因产物是双亮氨酸拉链激酶(dlk)、亮氨酸拉链激酶(lzk)、atf2、jun、性别决定区y(sry)-盒11(sox11)、肌细胞增强因子2a(mef2a)、jnk1-3、mkk4、mkk7、sox11或puma、或其组合。
在一些实施方案中,一种或多种基因产物是dlk/lzk、mkk4/7、jnk1/2/3或sox11/atf2/jun/mef2a途径的成员。
在一些实施方案中,靶序列选自seqidno:143-163中所示的核苷酸序列、或其组合。
在一些实施方案中,对主体的施用通过植入、注射或病毒而发生。
在一些实施方案中,对主体的施用通过视网膜下注射而发生。
在一些实施方案中,主体是人。
本发明的另一个方面涉及改变细胞中的一种或多种基因产物表达的方法,其中所述细胞包含编码一种或多种基因产物的dna分子,所述方法包括向所述细胞内引入包含一种或多种载体的非天然存在的核酸酶系统,所述载体包含:a)与编码核酸酶系统指导rna(grna)的至少一种核苷酸序列可操作地连接的启动子,其中所述grna与dna分子的靶序列杂交;以及b)在细胞中可操作的调节元件,其与编码基因组靶向核酸酶的核苷酸序列可操作地连接,
其中组件(a)和(b)位于系统的相同或不同载体上,其中所述grna靶向所述靶序列且与之杂交,并且核酸酶切割dna分子以改变一种或多种基因产物的表达。
在一些实施方案中,该系统是crispr。
在一些实施方案中,将系统包装到单个腺伴随病毒(aav)颗粒内。
在一些实施方案中,该系统使一种或多种基因产物失活。
在一些实施方案中,核酸酶系统切除至少一个基因突变。
在一些实施方案中,启动子是双向启动子。
在一些实施方案中,双向启动子是h1。h1启动子既是polii又是poliii启动子。
在一些实施方案中,h1启动子包含:a)提供在编码grna的至少一种核苷酸序列的一个方向上的转录的控制元件;以及b)提供在编码基因组靶向核酸酶的核苷酸序列的相反方向上的转录的控制元件。
在一些实施方案中,基因组靶向的核酸酶是cas9。
在一些实施方案中,cas9蛋白进行密码子优化用于在细胞中表达。
在一些实施方案中,启动子与至少一个、两个、三个、四个、五个、六个、七个、八个、九个或十个grna可操作地连接。
在一些实施方案中,细胞是真核或非真核细胞。
在一些实施方案中,真核细胞是哺乳动物细胞或人细胞。
在一些实施方案中,细胞是视网膜感光细胞。
在一些实施方案中,细胞是视网膜神经节细胞。
在一些实施方案中,一种或多种基因产物是lca10cep290。
在一些实施方案中,靶序列选自seqidno:1-109、164-356、735-738中所示的核苷酸序列,或其组合。
在一些实施方案中,靶序列包含可操作地连接的seqidno:1、2、3和4。
在一些实施方案中,载体包含seqidno:110中所示的核苷酸序列。
在一些实施方案中,一种或多种基因产物是视紫红质。
在一些实施方案中,靶序列是视紫红质基因中的突变。
在一些实施方案中,靶序列是在视紫红质基因的r135处的突变。
在一些实施方案中,在r135处的突变选自r135g、r135w、r135l。
在一些实施方案中,靶序列选自seqidno:111-126中所示的核苷酸序列、或其组合。
在一些实施方案中,grna序列选自seqidno:127-142中所示的核苷酸序列、或其组合。
在一些实施方案中,一种或多种基因产物是双亮氨酸拉链激酶(dlk)、亮氨酸拉链激酶(lzk)、atf2、jun、性别决定区y(sry)-盒11(sox11)、肌细胞增强因子2a(mef2a)、jnk1-3、mkk4、mkk7、sox11或puma、或其组合。
在一些实施方案中,一种或多种基因产物是dlk/lzk、mkk4/7、jnk1/2/3或sox11/atf2/jun/mef2a途径的成员。
在一些实施方案中,靶序列选自seqidno:143-163中所示的核苷酸序列、或其组合。
在一些实施方案中,一种或多种基因产物的表达降低。
已在上文中陈述了本文公开主题的某些方面,其全部或部分地由本文公开主题解决,当与如本文下文更佳描述的所附实施例和附图结合进行描述时,其他方面将变得显而易见。
附图简述
因此已经概括地描述了本文公开主题,现在将参考附图,所述附图不一定按比例绘制,并且其中:
图1显示了平台技术。h1双向启动子的基因组序列,其中polii转录物以蓝色显示,并且poliii转录物以橙色显示(左)。在单个aav载体内包装spcas9和grna(右)。
图2显示了aav-h1-crispr对小鼠视网膜的递送。经改造以表达gfp代替cas9的病毒证实了在视网膜下注射后,在外核层(onl)中的小鼠光感受器的既有效又特异性的转导。这些是受lca突变影响的细胞。
图3显示了cas9切口酶方法的图示,其需要在相反链上的两个紧密相对的靶位点(l和r)。
图4显示了lca10突变。四个鉴定的grna位点将导致~1kb缺失,从cep290中去除隐藏外显子x。
图5显示了以4550bp递送2个grna的目前sacas9方法(左)相对于使用4335bp递送4个grna的紧凑h1系统(右)。aav包装容量通过虚线指示。
图6显示了所有sacas9位点(cep290突变的上游1kb和下游1kb)。
图7显示了可用于靶向的sacas9位点(全部以5'g开始)。
图8显示了安全得多的sacas9切口酶方法。
图9显示了sacas9切口酶缺失(1078bp)。
图10显示了潜在的spcas9位点。
图11显示了在cep290(lca10)靶向区域中,通过5'核苷酸的sacas9或spcas9的crispr位点数目。
图12显示了具有四个grna的克隆的~4.2kbsacas9构建体。
图13显示了视紫红质基因结构。
图14显示了全世界rho基因的突变谱(来自http://www.hindawi.com/journals/bmri/2014/302487/)。
图15显示了视紫红质arg135突变。
图16显示了2个法**族的r135w家谱(来自audoi等人investophthalmolvissci.(2010)jul;51(7):3687-700)。
图17显示了来自5代西西里人家谱的r135w(来自pannaralemr等人ophthalmology.(1996)sep;103(9):1443-52)。
图18显示了具有r135l的六代瑞典家族(来自andréassons等人ophthalmicpaediatrgenet.(1992)sep;13(3):145-53)。
图19显示了致敏的激酶组筛选将lzk鉴定为与dlk合作以促进rgc细胞死亡。
图20显示了全基因组sirna筛选将atf2、sox11和mef2a鉴定为rgc细胞死亡的介质。
图21显示了lzk/dlk依赖性rgc细胞死亡的下游介质。
图22显示了lzk中的钙感知基序对毒性是可有可无的。
图23显示了锤头状核酶-sgrna融合物以增加可靶向的spcas9位点的数目。
图24显示了基于网络的sirna和sipool筛选具有改善的灵敏度和特异性。
图25显示了h1启动子允许polii和poliii转录物的双向表达。
图26显示了基于流式细胞术的rgc定量。
图27显示了体外dlk外显子1(a)和外显子2(b)的crispr靶向。dlk的外显子1和外显子2,其中靶位点以蓝色显示,并且t7e1引物以绿色显示。
图28包括两个小图a和b,显示了体外dlk的crispr靶向。图28a显示了筛选grna靶向来自小鼠的dlk基因的能力。靶位点命名法根据http://crispr.technology。图28b显示了使用双向启动子表达cas9的体外切割,并且grna证实了dlk基因座的有效靶向。对照是cas9和grna的标准2-质粒转染。图28b显示了测试从h1双向启动子驱动cas9和grna两者的能力的实验。使用h1双向启动子,用标准两个质粒(cas9和grna)或单个质粒转染培养中的细胞。t7ei测定指示使用任一系统的可比较水平的切割。
图29显示了在体外通过aav的dlk靶向。
图30包括三个小图a、b和c,显示了体内rgc中的双向表达。图30a显示了包装到aav内的构建体。图30b显示了体内gfp的细胞类型表达受所用aav血清型影响。上图显示了光感受器中的优先表达,并且下图显示了rgc中的优先表达。h1启动子在由aav5递送的感光细胞或由aav2(对照)递送的视网膜神经节细胞中明确地表达gfp。通过视网膜下注射将两种病毒均递送至p0.5天小鼠。两者均通过图30a中所示的报道分子的视网膜下注射来递送。图30c显示了在报道构建体的aav2玻璃体内递送15天后,通过铺片(flatmount)的gfp表达。
图31包括三个小图a、b和c,显示了使用自互补的aav病毒的双向靶向。图31a显示了自互补的aav构建体,其表达来自双向启动子的核mcherry和grna。该图进一步显示了测试使用自互补的aav(scaav)递送grna和荧光报告蛋白(h2b-mcherry)的能力的实验。从cas9小鼠收获细胞,并且在体外转导。scaav的益处是快得多地测试构建体的能力,因为表达在几天而不是几周内发生。图31a显示了使用h1启动子生成构建体,以同时表达grna(以黑色显示)和mcherry。grna靶向dlk小鼠基因,该基因在失活时导致增强的视网膜神经节细胞存活。包装构建体以产生自互补的aav(scaav)。从cas9转基因小鼠(其共表达gfp)收获视网膜神经节细胞,并且用scaav病毒转导;mcherry表达在表达gfp的所有细胞中都是明显的,指示来自构建体的高效转导和表达。图31a还显示了测试使用自互补的aav(scaav)递送grna和荧光报告蛋白(h2b-mcherry)的能力。从cas9小鼠收获细胞,并且在体外转导。scaav的益处是快得多地测试构建体的能力,因为表达在几天而不是几周内发生。图31b显示了体外转导rgc的scaav报道基因的体外表达;gfp表达来自cas9小鼠。图31c显示了如通过bglii测定检测的高效靶向(基本上)。通过ssaav递送grna(mm079)并且从小鼠呈现cas9。
图32包括五个小图a-e,显示了使用自互补的aav病毒的双向靶向。转导衍生自cas9小鼠的wtrgc或rgc的scaav病毒的滴定。图32c显示了当存在cas9时,在rgc中发生基因组编辑。这些体外实验证实非常高水平(~)的切割。在该测定中,我们通过限制性酶位点的丧失来测定切割。在wt动物中,pcr产物被bglii完全消化,然而,在用aav转导的cas9小鼠中,在更高浓度下存在基本上无法检测的bglii切割,指示~crispr切割。图32d和32e显示了在治疗的眼中,在视神经挤压后,视网膜神经节细胞的体内挽救。使用h1启动子生成构建体,以同时表达grna(以黑色显示)和mcherry。grna靶向dlk小鼠基因,该基因在失活时导致增强的视网膜神经节细胞存活。包装构建体以产生自互补的aav(scaav)。将病毒玻璃体内施用到cas9转基因或作为对照的wt小鼠内。在视神经挤压后14天定量视网膜神经节细胞存活,指示与对照相比,crispr递送导致处理小鼠中的rgc存活(两只小鼠均接受crisprgrna,但小鼠之间的差异是cas9的存在,这是基因组编辑所必需的。)。
图33包括三个小图a、b和c,显示了dlk的crispr靶向导致rgc存活。通过慢病毒转导来自cas9小鼠的rgc导致如通过bglii测定测量的~切割。dlk的破坏导致rgc存活中的显著增加,证实dlk靶向作为视神经病变中的治疗靶的可能性。
图34包括两个小图a和b,显示了体外的lzk的crispr靶向。图34a显示了lzk的外显子1,其中靶位点以蓝色显示,并且t7e1引物以绿色显示。图34b显示了lzk的外显子2,其中靶位点以蓝色显示,并且t7e1引物以绿色显示。
图35包括七个小图a-h,显示了激酶组的致敏sirna筛选将lzk鉴定为体外rgc细胞死亡的介质。图35a显示了用cre表达或对照腺病毒转导并且在陶扎色替(1μm)或媒介物对照的存在下培养的dlkfl/flrgc的存活。图35b描绘了显示激酶组文库中所有1,869个sirna(在dlksirna的存在下转染)的标准化存活的直方图。箭头显示了关于lzk的三种sirna中的每一种的存活。图35c显示了用与四种独立的lzksirna之一或非靶向对照组合的对照或dlksirna转染的wtrgc的存活。图35d显示了在用对照、dlk、lzk或dlk和lzksipool两者转染后,wtrgc的基于毛细管的免疫测定。图35e显示了用渐增量的对照、dlk、lzk或dlk和lzksipool两者转染的wtrgc的存活。图35f显示了用dlksirna和对照sirna或靶向激酶的混合谱系激酶家族的其他成员的四种独立sirna之一转染的wtrgc的存活。图35g显示了用对照、dlk、lzk或dlk和lzksipool两者转染,并且在递增剂量的陶扎色替的存在下培养的wtrgc的存活。图35h描绘了在秋水仙碱(1μm)添加后两天,在神经营养因子(nt,50ng/mlbdnf、5ng/mlgdnf、5ng/mlcntf)的存在或不存在下,用sipool转染的wtrgc的存活(±sd)。*p<0.05,曼惠二氏u检验。d/l,dlk/lzk。
图36包括六个小图a和f,显示了具有dlk和lzk的靶向缺失的rgc在体外和体内对轴突损伤诱导的细胞死亡是高度抗性的。图36a显示了用于生成组成性和条件性lzk敲除小鼠的方法的图解。插图显示了dna印迹,其确认杂合动物中单一靶向构建体的存在。图36b显示了在免疫淘选损伤后0或24小时,从wt相对于lzk-/-小鼠中分离的rgc的基于毛细管的免疫测定(上图)和定量(下图)。图36c显示了在视神经挤压或假手术对照后两周,针对未受伤的对照标准化的,存活rgc的数目的基于流式细胞术的定量。ns,不显著的,曼惠二氏u检验。图36d显示了从wt或lzkfl/fl小鼠中分离并且用cre表达或对照腺病毒转导的rgc的基于毛细管的免疫测定(上图)和定量(下图)。图36e显示了用增加量的cre表达或对照腺病毒转导的存活wt、dlkfl/fl、lzkfl/fl或dlkfl/fllzkfl/flrgc。图36f显示了在视神经挤压或假手术对照后两周,针对未受伤的对照标准化的,存活rgc的数目的基于流式细胞术的定量。在手术前两周,所有眼都注射了109vgaav2-cre。*p<0.05,曼惠二氏u检验。
图37包括五个小图a-e,显示了lzk激酶信号传导触发经由mkk4/7和jnk1-3激酶级联的rgc细胞死亡。图37a显示了wtrgc的存活,所述wtrgc用dlk/lzksipool转染并然后通过用表达wt或突变体、sipool抗性、人lzkcdna的腺病毒转导而用lzk信号传导重构。图37b-c显示了用渐增量的cre表达或对照腺病毒转导的wt(b-c)、jnk1fl/fljnk2-/-jnk3-/-(b)或mkk4fl/flmkk7fl/fl(c)rgc的存活。图37d-e显示了wt(d-e)、jnk1fl/fljnk2-/-jnk3-/-(d)或mkk4fl/flmkk7fl/fl(e)rgc的存活,其用dlk/lzksipool转染,用cre表达或对照腺病毒转导,然后两天后,通过用表达人lzkcdna的或对照腺病毒转导重构lzk信号传导。
图38包括三个小图a-c,显示了全基因组sirna筛选将atf2、puma和mef2a鉴定为rgc细胞死亡的介质。图38a描绘了直方图,其显示了靶向全基因组文库中17,575个基因中的每一个的中值存活促进sirna的标准化、种子调整的存活。箭头显示了靶向atf2、puma和mef2a的中值存活促进sirna的存活。图38b描绘了用靶向atf2、puma或mef2a的四种独立sirna之一或者非靶向对照转染的wtrgc的存活。虚线指示存活阈值大于来自阴性对照的3sd。图38c显示了用渐增量的对照和atf2(左)、puma(中)或mef2a(右)sipool转染的wtrgc的存活。
图39包括七个小图a-g,显示了具有atf2和mef2a的转录调节结构域的靶向破坏的rgc对体内轴突损伤诱导的细胞死亡是部分抗性的。图39a显示了用dlk/lzk或pumasirna转染,并且用wt或kd人lzk转导的wtrgc的存活。图39b显示了用cre表达或对照腺病毒转导的mef2afl/flrgc的基于毛细管的免疫测定。图39c显示了用渐增量的cre表达或对照腺病毒转导的wt或mef2afl/flrgc的存活。图39d显示了用cre表达或对照腺病毒转导mef2afl/fl相对于mef2afl/flmef2cfl/flmef2dfl/flrgc的存活中的倍数变化。图39e显示了在视神经挤压或假手术对照后两周,针对未受伤的对照标准化的,存活rgc的数目的基于流式细胞术的定量。在手术前两周,所有眼都注射了109vgaav2-cre。*p<0.05,曼惠二氏u检验。图39f显示了在视神经挤压或假手术对照后两周,作为总数的百分比表示的tubb3/p-s408mef2a数目的基于流式细胞术的定量。图39g显示了用渐增量的cre表达或对照腺病毒转导的wt或atf2fl/flrgc的存活。
图40包括六个小图a-g,显示了致敏的全基因组sirna筛选将jun和sox11鉴定为rgc细胞死亡的介质。图40a描绘了直方图,其显示了靶向全基因组文库中的每种基因(在lzksipool的存在下转染)的sirna微型库(minipool)的标准化存活。箭头显示了靶向dlk的顶部sirna微型库的存活。图40b显示了在免疫淘选损伤后的指示时间的wtrgc中针对gapdh水平标准化,并且用对照sipool或者针对dlk/lzk或sox11的sipool转染的sox11mrna的定量pcr(qpcr)测定。图40c显示了用渐增量的lzk和/或sox11sipool转染的wtrgc的存活。图40d显示了用lzk或lzk/sox11sipool转染,并且通过用对照或表达人sox11cdna的腺病毒转导对于sox11信号传导重构的wtrgc的存活。图40e显示了用渐增量的cre表达或对照腺病毒转导的wt或sox11fl/flrgc的存活。图40f显示了在视神经挤压或假手术对照后两周,针对未受伤的对照标准化的,存活rgc的数目的基于流式细胞术的定量。在手术前两周,所有眼都注射了109vgaav2-cre。*p<0.05,曼惠二氏u检验。图40g描绘了在用腺病毒转导的sox11fl/flrgc中,针对gapdh水平标准化的sox11mrna的qpcr测定。
图41包括七个小图a-h,显示了dlk/lzk依赖性细胞死亡由一组四种转录因子介导:jun、atf2、sox11和mef2a。图41a显示了用指示的sipool转染的wtrgc的存活。图41b显示了用dlk/lzksipool和对照或jun/atf2/sox11/mef2asipool转染,然后通过用sipool抗性、人lzkcdna表达的或对照腺病毒转导,用lzk信号传导重构的wtrgc的存活。图41c显示了用lzksipool和靶向dlk的tracrrna或sgrna转染的wt或spcas9敲入rgc的存活。图41d显示了用dlksipool和靶向lzk的tracrrna或sgrna转染的wt或spcas9敲入rgc的存活。图41e显示了用各个sgrna或靶向dlk和/或lzk的sgrna库转染的wt或spcas9敲入rgc的存活。图41f显示了通过转染渐增量的靶向单独或组合的四种转录因子(jun、atf2、sox11、mef2a)中的每一种的sgrna赋予标准化的存活(spcas9-wt),并且与用阴性对照tracrrna或靶向dlk/lzk的阳性对照sgrna的转染进行比较。图41h描绘了在腺病毒转导以激活lzk信号传导后两天,用sgrna转染的spcas9rgc的存活(±sd)。*p<0.05曼惠二氏u检验比较dlk/lzk和转录因子sgrna。图41g描绘了显示对于轴突损伤后的rgc细胞死亡所提出的途径的图解。
图42(与图61相关)含有6个小图a-f,显示了lzk是原代rgc中细胞死亡的介质。
图43(与图62相关)含有5个小图a-e,显示了定量rgc存活的基于流式细胞术的测定的开发。
图44(与图63相关)含有4个图a-d,显示了全基因组sirna筛选结果。
图45(与图64和65相关)含有8个小图a-h,显示了mef2a、puma和atf2的敲低,并且视神经损伤导致dlk依赖性mef2a磷酸化。
图46(与图66相关)含有5个小图a-e,显示了全基因组、致敏的sirna筛选结果。
图47(与图68相关)含有2个小图a-b,显示了在原代rgc中dlk的crispr敲除。
图48含有两个小图a-b,描绘了存活图。
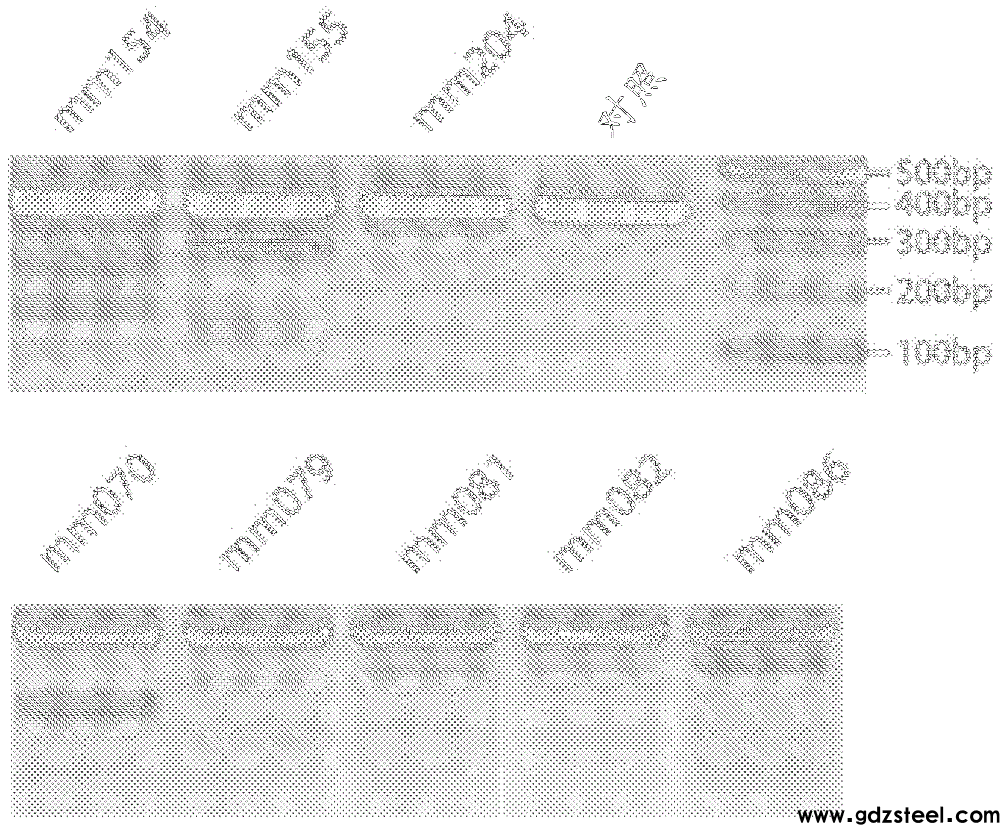
图49描绘了示踪剂rna,mm190、mm204、mm094、mm079、mm936、mm926、mm375、mm775、mm878和mm942的表。
图50显示了用于靶向ceP20的aav2构建体大小。构建体大小为4,781bp。启动子是h1双向(小鼠)。polii终止子是spa,且aav血清型是aav2。
图51显示了lca10由cep290中的内含子突变引起。描绘了cep290基因。ivs26c.2991+1655a>g突变导致异常剪接以及128bp隐藏外显子x的包括(下图)。
图52描绘了h1rna和parp-2基因座的基因组组构。上面显示的是按比例描绘的向右侧转录的parp-2基因(蓝色)和转录到左侧的h1rna基因(橙色)的描述。下面是关于两个基因的启动子区的放大区域。
图53描绘了spcas9靶位点和lca10突变。a>g突变的定位在具有外显子x(橙色)的3'末端和spcas9靶位点(蓝色)的背景下指示。spcas9切割点由两个箭头描绘,并且用于剪接的关键核苷酸是加框的。
图54描绘了高保真/高特异性spcas9变体。espcas9点突变以蓝色指示,并且spcas9-hf1点突变以橙色指示。
图55描绘了cas9切口酶方法。切口酶需要在相反链上的两个紧密相对的靶位点(l和r;上图),以生成双链dna断裂(下图)。
图56包括三个小图a-c,描绘了lca10crispr/aav治疗剂。图56a描绘了aav病毒和包装容量的卡通图(cartoon)。图56b描绘了关于spcas9靶向构建体的图解,其包括来自sa1的espcas9和spcas9-hf变体。图56c描绘了具有如sa2中所述的四种grna的sacas9切口酶的图解。
图57含有八个小图,a-h。图57a和57b描绘了cep290的基因组基因座,其中指示了深层内含子lca10突变(a->g)的定位。这种突变导致隐藏外显子(外显子x)包括在mrna内,导致截短的蛋白质。a->g突变可以通过落在突变序列上的crispr-cas9位点靶向。预期通过这种grna的靶向导致正确的剪接,因为在剪接点周围形成的插入缺失可以损害该伪外显子的剪接。下图显示了将dsdna断裂靶向内含子区域以恢复正常cep290表达的一般策略。图57c描绘了正常的cep290基因(外显子25–28)。图57d描绘了点突变,其随后导致隐藏外显子包括在cep290内。图57e和57f描绘了报告的尝试使用sacas9(因为它足够小以纳入aav内)去除点突变序列的策略。该策略寻求去除内含子的大区段,以去除点突变序列。这很可能是因为在点突变附近不存在sacas9位点。使用sacas9的这种策略可能具有两种后果:(1)效率较低,因为需要两次dna切割而不是一次;(2)安全性问题例如染色体易位或重排,以及发生插入诱变的可能性,以及(3)更多脱靶的可能性。相反,使用双向策略,可以通过aav递送cas9(或cas9变体,或cpf1或cpf1变体),开放了更多的靶向位点,以及恢复关于cep290的正常剪接的策略。图57g和57h描绘了cep290靶向位点的切割,如通过质粒转染或通过aav病毒的转导后对bmri切割的抗性所测量的。
图58描绘显示了用于在体内靶向视紫红质的aav5构建体大小。构建体大小为4,996bp。启动子是h1双向(人)。polii终止子是sv40。aav血清型是aav5。
图59含有五个小图a-e,显示了玻璃体内注射后在体内的双向表达。图59a描述了体内h1双向启动子构建体的验证。如图59a上所示,使用h1启动子生成的构建体用于同时表达rna(以黑色显示grna)和gfp。gfp用于提供来自h1启动子的polii表达的视觉读出。然后使用aav2*(y444f+y500f+y730f三重衣壳突变,由*指示),将侧翼为aavitr序列的构建体包装到病毒内,所述aav2*具有对于视网膜神经节细胞转导的优先。通过玻璃体内注射递送病毒;使用单独的媒介物的对照注射递送至对照眼。注射后14天,将小鼠镇静并使用microiii视网膜成像显微镜显现gfp表达。可以从活小鼠中检测到弥漫的gfp表达,如左图上所示。图59c显示没有gpf表达。对小鼠实施安乐死,并且从视网膜铺片显现gfp表达,指示视网膜神经节细胞层中的gfp表达,如使用aav2*血清型将预期的。图59c显示了h1双向启动子用于表达gfp(和空grna),并且包装到aav2或aav5病毒内。该实验用于测试不同细胞类型中的h1表达并且验证细胞类型的向性。图59d显示了描绘血清型和向性的含义的卡通图示。血清型指aav的不同“毒株”,并且向性指给定毒株可以感染的细胞类型。该特性提供了另外的安全层,因为特定的血清型可以用于靶向所需的细胞类型。例如,在视网膜中,aav血清型得到充分表征,并且可以用于优先感染某些细胞,即使它们被许多不同的细胞类型包围。特别地,aav5证实光感受器特异性。
图60含有两个小图a-b,显示了在体内通过crispr的视网膜基因组编辑和使用snp靶向显性等位基因的策略。该实例用于靶向视紫红质中的p23h突变。图60b显示了体内显性突变(rhop23h)的crispr靶向,其描绘了snp用于等位基因特异性和顺式失活的用途,以及改造的cas9变体靶向p23h的用途。在左侧上显示了使用与野生型近交小鼠品系cast/eij杂交的c57bl品系上的p23h纯合小鼠的育种方案。如通过测序所示,cast品系携带其他品系中未携带的非同义snp。该snp不改变wt视紫红质蛋白质序列,并且可以用于区分wt视紫红质序列和p23h序列。p23h点突变(c->a)落在来自cas9pam序列的ngg的n上,并且因此grna将同等地靶向wt和p23h序列两者。cast序列携带另外的碱基变化,其允许grna靶向突变而不是wt序列。
图61(与图42相关)含有11个小图a-k,显示了lzk是原代rgc中的细胞死亡的介质。图61a显示了在铺平板时,rgc的celltiter-glo(“存活(rlu)”)和基于细胞组学的(“活rgc”)定量的比较。图61b-c显示了在用sirna(b)或sipool(c)转染后,rgc中lzk的基于毛细管的免疫测定(上图)和定量(下图)。图61d显示了在秋水仙碱后48小时,用lzksipool±dlksipool转染的rgc的celltiter-glo(“存活(rlu)”)和基于细胞组学的(“活rgc”)定量的比较。ns,通过曼惠二氏u检验不显著的。图61e显示了通过自动荧光显微镜检查定量的活rgc(钙黄绿素-am-染色/乙啡啶同型二聚体-排除;±sd)。图61f显示了在用对照或lzksipool转染,并且用表达小鼠sirna抗性、人lzkcdna或gfp对照的腺病毒转导后**,rgc中lzk的基于毛细管的免疫测定(上图)和定量(下图)。图61g显示了用表达小鼠sirna抗性、人lzkcdna或gfp对照的腺病毒重构lzk信号传导后两天,用dlk/lzksipool转染的wtrgc的存活(±sd)(“lzk重构测定”)。图61h-i显示了用sipool转染,并且在免疫淘选损伤后三天用钙黄绿素-am染色的rgc中的神经突长度(每个神经元的平均值)的代表性显微照片(h)和定量(i)。图61j显示了在秋水仙碱攻击后两天,用腺病毒转导的wtrgc的存活(±sd)。图61k显示了在免疫淘选损伤后**,来自wtrgc的免疫共沉淀测定。
图62(与图43相关)含有6个小图a-f,显示了定量rgc存活的基于流式细胞术的测定的开发。图62a描绘了来自未受伤的野生型c57bl/6小鼠的代表性视网膜铺片的免疫荧光染色。图62b描绘了在免疫淘选后立即通过fc分析的免疫淘选的p0-3小鼠rgc的2d图。图62c-d描绘了通过fc分析的未受伤的视网膜(c)或onc后两周(d)的代表性2d图。(c)中的门(gates)用于显示对于thy1.2/neun是双重阳性的tubb3/sncg双重阳性细胞的百分比,或反之亦然,以便分别估计该技术的特异性和灵敏度。平均值显示于下文。图62e描绘了在onc或假手术后的各个时间点(括号中的n),存活rgc的基于fc的定量(±sd)。图62f描绘了存活rgc的基于fc的定量(±sd)相对于免疫染色铺片的手动计数的比较。在两种情况下,通过sncg/tubb3双重染色鉴定rgc。ns,通过曼惠二氏u检验不显著的。
图63(与图44相关)含有四个小图a-d。全基因组sirna筛选结果。图63a-b显示了对如通过通过种子序列的贡献校正后通过z得分排列的**48种基因(a)或如通过haystack分析确定的**14种基因(b)的等级次序。验证的基因以红色显示。图63c-d描绘了二次筛选的结果,其中用四种独立的sirna转染rgc,所述sirna靶向通过种子校正的(c)或haystack(d)分析提名的**基因(topgenes)。虚线显示了存活阈值大于来自阴性对照的3sd。
图64(与图45相关)含有五个小图a-e,显示了mef2a、puma和atf2的敲低。图64a-b显示了基于毛细管的免疫测定,其中相关的定量(上图、中图)或qpcr(下图)对用各个sirna(a)或sipool(b)转染的rgc执行。图64c显示了来自用腺病毒转导三天的atf2fl/flrgc的atf2的基于毛细管的免疫测定。图64d显示了在秋水仙碱攻击后两天,用腺病毒转导的rgc的存活(±sd)。图64e显示了在onc或假手术后两周,针对未受伤的对照标准化的,存活rgc的基于fc的定量(±sd)。在手术前两周,所有眼都注射了109vgaav2-cre。*p<0.05,曼惠二氏u检验。
图65(与图45相关)含有两个小图a和b,显示了视神经损伤导致dlk依赖性mef2a磷酸化。图65a显示了在onc或假手术后两天,代表性视网膜切片的合并免疫荧光染色。图65b显示了在onc或假手术后两天,通过fc分析的代表性wt或dlkfl/fl视网膜的2d图。
图66(与图46相关)含有五个小图a-e,显示了全基因组、致敏的sirna筛选结果。图66a显示了在校正通过种子序列的贡献后,通过z得分排列的**48种候选基因的等级次序。先前验证的基因以红色显示。图66b显示了散点图,其显示了关于全基因组文库中每个微型库的种子校正活性。先前验证的基因以红色显示。图66c显示了二次筛选的结果,其中用四种独立的sirna转染rgc,所述sirna靶向通过种子校正的分析提名的**基因。虚线显示了存活阈值大于来自阴性对照的3sd。图66d显示了如通过haystack分析确定的**基因的等级次序。图66e显示了二次筛选的结果,其中用四种独立的sirna转染rgc,所述sirna靶向通过haystack分析提名的**基因。虚线显示了存活阈值大于来自阴性对照的3sd。
图67显示了转录因子对神经突向外生长的作用。用sipool转染,并且在免疫淘选损伤后三天用钙黄绿素-am染色的rgc中的神经突长度(每个神经元的平均值;±sd)的定量。*p<0.05曼惠二氏u检验。
图68(与图47相关)含有四个小图a-d,显示了在原代rgc中dlk的crispr敲除。图68a显示了在用tracrrna或dlkgrna#4转染后三天,从spcas9rgc基因组dna扩增,并且在dlk/lzk抑制剂的存在下维持以避免选择的pcr产物的bglii消化物。pcr区域包括dlkgrna#4靶位点或由http://crispr.mit.edu预测的前14个脱靶(所有这些都维持bglii位点)。图68b显示了亚克隆后dlk#4pcr产物的测序结果。图68c显示了在秋水仙碱攻击后两天,在神经营养因子(nt,50ng/mlbdnf、5ng/mlgdnf、5ng/mlcntf)的不存在或存在下,用靶向dlk和lzk的tracrrna或sgrna转染的wt相对于表达spcas9的rgc(±sd)的存活。*p<0.05,曼惠二氏u检验。图68d显示了通过转染第二组sgrna而赋予的存活中的差异(spcas9-wt;±sd),所述sgrna靶向单独或组合的四种转录因子中的每一种,并且与阴性对照tracrrna进行比较。
图69描绘了包括通过舒尼替尼(fda批准的小分子抑制剂)的dlk和lzk的药理学抑制,促进人esc衍生的rgc的存活。图69a描绘了在dlk/lzk抑制剂陶扎色替(1μm)、genentech抑制剂123(0.1μm)或媒介物对照的存在下,用媒介物或秋水仙碱(1μm)攻击后两天,hesc衍生的rgc的存活(±sd)。图69b描绘了在渐增剂量的舒尼替尼的存在下,用秋水仙碱(1μm)攻击后两天,hesc衍生的rgc的存活(±sd)。
图70描绘了使用靶向视紫红质的构建体的一个实施方案的体内基因组编辑。
图71描绘了使用靶向视紫红质的构建体的一个实施方案的体内基因组编辑。来自总视网膜的细胞用于测定切割(未纯化的光感受器),并且轻微的切割水平在14天时可检测到,其在28天时增加。
图72描绘了采用核酸酶死亡形式的cas9用于调节转录调节的融合构建体。这些构建体可以使用h1双向启动子通过aav递送。
图73显示了启动子区中的视紫红质启动子和蛋白质结合位点的示意图。通过破坏这些相互作用,可以顺式阻遏等位基因的转录,并且通过利用启动子区中的snp,可以递送非切割形式的cas9以治疗adrp。
图74描绘了视紫红质启动子和rer区域的示意图(上图)。中图显示了来自三种不同小鼠细胞/品系的pcr产物,其是sanger测序的。所鉴定的snps显示在底部图解上的线。我们已鉴定了几个区域,其允许我们利用这些序列变异来顺式调节视紫红质的表达。
图75描绘了关于crispr靶向位点,在视紫红质启动子区(从rer到转录起始)中的~2kb区域的扫描。我们选择在rer附近的6个位点和在近端启动子区附近的6个位点,并且测定它们的切割(我们使用切割效率作为用于鉴定允许高结合效率的可接近区域的代理)。我们已鉴定了几种候选序列,其可以通过简单破坏rer/近端启动子区之间的染色质环,或通过使用dcas9融合(激活物/阻遏物结构域),允许通过dcas9的调节。
图76描绘了用于测试对部分人序列的切割和非切割方法的rho-gfp小鼠。下文是使用来自rho-gfp小鼠的纯化的光感受器并且通过crispr靶向寻找gfp丧失的体外测定。
图77描绘了用于测试与激活物或阻遏物结构域融合的核酸酶死亡的cas9构建体的报告测定。
发明详述
本文公开主题现在将参考附图在下文中更全面地描述,所述附图中显示了本文公开主题的一些但并非全部实施方案。相同的数目自始至终指相同的要素。本文公开主题可以以许多不同的形式体现,并且不应被解释为限于本文阐述的实施方案;相反,提供这些实施方案使得本公开内容满足适用的法律要求。实际上,本文公开主题所属领域的技术人员将会想到本文阐述的本文公开主题的许多修改和其他实施方案,其具有前述描述和相关附图中呈现的教导的益处。因此,应理解,本文公开主题并不限于所公开的特定实施方案,并且修改和其他实施方案预期包括在所附权利要求的范围内。
基因组编辑技术,例如锌指核酸酶(zfn)(porteus和baltimore(2003)science300:763;miller等人(2007)nat.biotechnol.25:778-785;sander等人(2011)naturemethods8:67-69;wood等人(2011)science333:307)和类转录激活因子效应物核酸酶(talen)(wood等人(2011)science333:307;boch等人(2009)science326:1509-1512;moscou和bogdanove(2009)science326:1501;christian等人(2010)genetics186:757-761;miller等人(2011)nat.biotechnol.29:143-148;zhang等人(2011)nat.biotechnol.29:149-153;reyon等人(2012)nat.biotechnol.30:460-465)已赋予生成靶向基因组修饰的能力,并且提供了**校正疾病突变的潜力。虽然有效,但这些技术受到实际局限性的阻碍,因为zfn和talen对均需要对于给定的dna靶位点合成大而独特的识别蛋白。几个团体更近已报道了通过使用改造的ii型crispr/cas9系统的高效基因组编辑,所述系统克服了这些关键局限性(cong等人(2013)science339:819-823;jinek等人(2013)elife2:e00471;mali等人(2013)science339:823-826;cho等人(2013)nat.biotechnol.31:230-232;hwang等人(2013)nat.biotechnol.31:227-229)。与相对耗时且难以制造的zfn和talen不同,依赖与合成指导rna(grna)偶联的cas9蛋白的核酸酶活性的crispr构建体合成简单且快速,并且可以多重使用。然而,尽管它们的合成相对容易,但crispr具有与其接近可靶向基因组空间相关的技术局限性,这是cas9本身的特性和其grna的合成两者的函数。
通过crispr系统的切割需要grna与20核苷酸的dna序列和必要的前间区序列邻近基序(pam)的互补碱基配对,所述pam是在靶位点3'发现的短核苷酸基序(jinek等人(2012)science337:816-821)。理论上,可以使用crispr技术靶向基因组中的任何独特的n20-pam序列。pam序列的dna结合特异性(其取决于所采用的特定cas9的起源物种而变化)提供了一个约束。目前,限制性更小且更常用的cas9蛋白来自化脓性链球菌(s.pyogenes),其识别序列ngg,并且因此,可以靶向基因组中任何独特的21核苷酸序列,随后为两个鸟苷核苷酸(n20ngg)。由蛋白质组分施加的可用靶向空间的扩展限于具有改变的pam要求的新型cas9蛋白的发现和使用(cong等人(2013)science339:819-823;hou等人(2013)proc.natl.acad.sci.u.s.a.,110(39):15644-9),或等待经由诱变或定向进化生成新型cas9变体。crispr系统的第二个技术约束来自于在5'鸟苷核苷酸处起始的grna表达。iii型rna聚合酶iii启动子的使用已特别顺应grna表达,因为这些短的非编码转录物具有明确的末端,并且转录的所有必需元件,其中排除了1+核苷酸,包含在上游启动子区中。然而,因为常用的u6启动子需要鸟苷核苷酸来起始转录,所以u6启动子的使用已进一步约束了gn19ngg的基因组靶向位点(mali等人(2013)science339:823-826;ding等人(2013)cellstemcell12:393-394)。替代方法,例如通过t7、t3或sp6启动子的体外转录,也需要起始鸟苷核苷酸(adhya等人(1981)proc.natl.acad.sci.u.s.a.78:147-151;melton等人(1984)nucleicacidsres.12:7035-7056;pleiss等人(1998)rna4:1313-1317)。
本文公开主题涉及靶向视网膜变性的crispr/cas9系统的修饰,其使用h1启动子来表达指导rna(grna或sgrna)(wo2015/19561,其通过引用整体并入本文),所述指导rna靶向视网膜变性相关基因,例如但不限于lca10cep290基因、视紫红质、双亮氨酸拉链激酶(dlk)、亮氨酸拉链激酶(lzk)、jnk1-3、mkk4、mkk7、atf2、jun、mef2a、sox11或puma。此类修饰的crispr/cas9系统可以以更高的功效、安全性和**度**地靶向这些视网膜变性中的致病性突变。此外,包含grna的这种修饰保留了crispr/cas9h1启动子系统的紧凑性质,其允许超过现有的crispr、talen或锌指技术的视网膜变性的更高分辨率靶向。
i.使用h1启动子靶向视网膜变性的crispr指导rna的表达
a.组合物
在一些实施方案中,用于治疗视网膜变性的本文公开方法利用包含先前在wo2015/195621(通过引用整体并入本文)中描述的非天然存在的crispr系统的修饰的组合物。此类修饰使用靶向视网膜变性相关基因的某些grna,所述基因例如但不限于lca10cep290基因、视紫红质、双亮氨酸拉链激酶(dlk)、亮氨酸拉链激酶(lzk)、jnk1-3、mkk4、mkk7、atf2、jun、mef2a、sox11或puma。在一些实施方案中,组合物包含(a)包含一种或多种载体的非天然存在的核酸酶系统(例如,crispr),所述载体包含:i)与编码核酸酶系统指导rna(grna)的至少一种核苷酸序列可操作地连接的启动子(例如,双向h1启动子),其中所述grna与主体的细胞中的dna分子的靶序列杂交,并且其中所述dna分子编码在细胞中表达的一种或多种基因产物;以及ii)在细胞中可操作的调节元件,其与编码基因组靶向核酸酶(例如cas9蛋白)的核苷酸序列可操作地连接,其中组件(i)和(ii)位于系统的相同或不同载体上,其中所述grna靶向所述靶序列且与之杂交,并且核酸酶切割dna分子以改变一种或多种基因产物的表达。在一些实施方案中,将系统包装到单个腺伴随病毒(aav)颗粒内。在一些实施方案中,腺伴随病毒(aav)可以包含51种人腺病毒血清型中的任一种(例如,血清型2、5或35)。在一些实施方案中,该系统使一种或多种基因产物失活。在一些实施方案中,核酸酶系统切除至少一个基因突变。在一些实施方案中,启动子包含:a)提供在编码grna的至少一种核苷酸序列的一个方向上的转录的控制元件;以及b)提供在编码基因组靶向核酸酶的核苷酸序列的相反方向上的转录的控制元件。在一些实施方案中,cas9蛋白进行密码子优化用于在细胞中表达。在一些实施方案中,启动子与至少一个、两个、三个、四个、五个、六个、七个、八个、九个或十个grna可操作地连接。在一些实施方案中,靶序列是cep290基因(例如,lca10cep290基因)中的突变。在一些实施方案中,关于cep290的靶序列选自seqidno:1-109、164-356、735-738中所示的核苷酸序列,或其组合。在一些实施方案中,靶序列包含可操作地连接的seqidno:1、2、3和4。在一些实施方案中,载体包含seqidno:110中所示的核苷酸序列。在一些实施方案中,一种或多种基因产物是视紫红质。在一些实施方案中,靶序列是视紫红质基因中的突变。在一些实施方案中,靶序列是在视紫红质基因的r135处的突变(例如,r135g、r135w、r135l)。在一些实施方案中,关于视紫红质r135的靶序列选自seqidno:111-126中所示的核苷酸序列、或其组合。在一些实施方案中,关于视紫红质r135的grna序列选自seqidno:127-142中所示的核苷酸序列、或其组合。在一些实施方案中,一种或多种基因产物是双亮氨酸拉链激酶(dlk)、亮氨酸拉链激酶(lzk)、jnk1-3、mkk4、mkk7、atf2、jun、mef2a、sox11或puma或其组合。在一些实施方案中,青光眼的突变靶包括但不限于optn、tbk1、tmco1、pmm2、gmds、gas7、fndc3b、txnrd2、atxn2、cav1/cav2、p16ink4a、six6、abca1、afap1和cdkn2b-as。在一些实施方案中,关于青光眼的靶序列选自seqidno:143-163中所示的核苷酸序列、或其组合。
在一些实施方案中,用于治疗视网膜变性的本文公开方法利用包含非天然存在的crispr系统的组合物,所述crispr系统包含一种或多种载体,所述载体包含:a)与编码crispr系统指导rna(grna)的至少一种核苷酸序列可操作地连接的h1启动子,其中所述grna与细胞中dna分子的靶序列杂交,并且其中所述dna分子编码在细胞中表达的一种或多种基因产物;以及b)在细胞中可操作的调节元件,其与编码cas9蛋白的核苷酸序列可操作地连接,其中组件(a)和(b)位于系统的相同或不同载体上,其中所述grna靶向所述靶序列且与之杂交,并且cas9蛋白切割dna分子以改变一种或多种基因产物的表达。
在一些实施方案中,用于治疗视网膜变性的本文公开方法利用包含非天然存在的crispr系统的组合物,所述crispr系统包含一种或多种载体,所述载体包含:a)与编码crispr系统指导rna(grna)的至少一种核苷酸序列可操作地连接的h1启动子,其中所述grna与真核细胞中dna分子的靶序列杂交,并且其中所述dna分子编码在真核细胞中表达的一种或多种基因产物;以及b)在真核细胞中可操作的调节元件,其与编码ii型cas9蛋白的核苷酸序列可操作地连接,其中组件(a)和(b)位于系统的相同或不同载体上,由此所述grna靶向所述靶序列且与之杂交,并且cas9蛋白切割dna分子,并且由此改变一种或多种基因产物的表达。在一个方面,靶序列可以是以任何核苷酸开始的靶序列,例如n20ngg。在一些实施方案中,靶序列包含核苷酸序列an19ngg。在一些实施方案中,靶序列包含核苷酸序列gn19ngg。在一些实施方案中,靶序列包含核苷酸序列cn19ngg。在一些实施方案中,靶序列包含核苷酸序列tn19ngg。在一些实施方案中,靶序列包含核苷酸序列an19ngg或gn19ngg。在另一个方面,cas9蛋白进行密码子优化用于在细胞中表达。在另一个方面,cas9蛋白进行密码子优化用于在真核细胞中表达。在一个进一步方面,真核细胞是哺乳动物细胞或人细胞。在另外一个方面,一种或多种基因产物的表达降低。
在一些实施方案中,用于治疗视网膜变性的本文公开方法利用包含非天然存在的crispr系统的组合物,所述crispr系统包含含有双向h1启动子的载体,其中所述双向h1启动子包含:a)提供在编码crispr系统指导rna(grna)的至少一种核苷酸序列的一个方向上的转录的控制元件,其中所述grna与真核细胞中dna分子的靶序列杂交,并且其中所述dna分子编码在真核细胞中表达的一种或多种基因产物;以及b)提供在编码ii型cas9蛋白的核苷酸序列的相反方向上的转录的控制元件,由此所述grna靶向所述靶序列且与之杂交,并且cas9蛋白切割dna分子,并且由此改变一种或多种基因产物的表达。在一个方面,靶序列可以是以任何核苷酸开始的靶序列,例如n20ngg。在一些实施方案中,靶序列包含核苷酸序列an19ngg。在一些实施方案中,靶序列包含核苷酸序列gn19ngg。在一些实施方案中,靶序列包含核苷酸序列cn19ngg。在一些实施方案中,靶序列包含核苷酸序列tn19ngg。在一些实施方案中,靶序列包含核苷酸序列an19ngg或gn19ngg。在另一个方面,cas9蛋白进行密码子优化用于在细胞中表达。在另一个方面,cas9蛋白进行密码子优化用于在真核细胞中表达。在一个进一步方面,真核细胞是哺乳动物细胞或人细胞。在另外一个方面,一种或多种基因产物的表达降低。
在一些实施方案中,crispr复合物包含足够强度的一个或多个核定位序列,以驱动crispr复合物在细胞(例如真核细胞)的核中以可检测的量的积累。不希望受理论束缚,认为核定位序列对于真核生物中的crispr复合物活性不是必需的,但包括此类序列增强了系统尤其是关于靶向核中的核酸分子的活性。在一些实施方案中,crispr酶是ii型crispr系统酶。在一些实施方案中,crispr酶是cas9酶。在一些实施方案中,cas9酶是肺炎链球菌(s.pneumoniae)、化脓性链球菌或嗜热链球菌(s.thermophilus)cas9,并且可以包括衍生自这些生物的突变的cas9。酶可以是cas9同源物或直向同源物。
一般而言,并且在本说明书自始至终,术语“载体”指能够转运它已与之连接的另一种核酸的核酸分子。载体包括但不限于其为单链、双链或部分双链的核酸分子;包含一个或多个游离末端,无游离末端(例如环状)的核酸分子;包含dna、rna或两者的核酸分子;以及本领域已知的其他各种各样的多核苷酸。一种类型的载体是“质粒”,其指环状双链dna环,另外的dna区段可以插入其内,例如通过标准分子克隆技术。另一种类型的载体是病毒载体,其中病毒衍生的dna或rna序列存在于载体中,用于包装到病毒(例如逆转录病毒、复制缺陷型逆转录病毒、腺病毒、复制缺陷型腺病毒和腺伴随病毒)内。病毒载体还包括由病毒携带用于转染到宿主细胞内的多核苷酸。
某些载体能够在它们引入其内的宿主细胞中自主复制(例如具有细菌复制起点的细菌载体和附加型哺乳动物载体)。其他载体(例如,非附加型哺乳动物载体)在引入宿主细胞内后整合到宿主细胞的基因组内,并且从而连同宿主基因组一起复制。此外,某些载体能够指导它们与之可操作地连接的基因的表达。此类载体在本文中称为“表达载体”。在重组dna技术中有用的常见表达载体经常以质粒的形式。
重组表达载体可以包含以适合于在宿主细胞中表达核酸的形式的本文公开主题的核酸,这意指重组表达载体包括一种或多种调节元件,其可以基于待用于表达的宿主细胞加以选择,其与待表达的核酸序列可操作地连接。
在重组表达载体内,“可操作地连接”预期意指目的核苷酸序列以允许核苷酸序列表达(例如在体外转录/翻译系统中,或当载体引入宿主细胞内时,在宿主细胞中)的方式与调节元件连接。
术语“调节元件”预期包括启动子、增强子、内部核糖体进入位点(ires)和其他表达控制元件(例如转录终止信号,例如多腺苷酸化信号和多聚u序列)。此类调节元件在例如goeddel(1990)geneexpressiontechnology:methodsinenzymology185,academicpress,sandiego,calif中描述。调节元件包括在许多类型的宿主细胞中指导核苷酸序列的组成型表达的那些元件,以及仅在某些宿主细胞中指导核苷酸序列的表达的那些元件(例如,组织特异性调节序列)。组织特异性启动子可以主要在所需目的组织,例如肌肉、神经元、骨、皮肤、血液、特定器官(例如肝、胰腺)或特定细胞类型(例如淋巴细胞)中指导表达。调节元件还可以以时间依赖性方式指导表达,例如以细胞周期依赖性或发育阶段依赖性方式,其可以是或可以不是组织或细胞类型特异性的。
在一些实施方案中,载体包含一种或多种poliii启动子、一种或多种polii启动子、一种或多种poli启动子或其组合。poliii启动子的实例包括但不限于u6和h1启动子。polii启动子的实例包括但不限于逆转录病毒劳氏肉瘤病毒(rsv)ltr启动子(任选地具有rsv增强子)、巨细胞病毒(cmv)启动子(任选地具有cmv增强子)(例如,boshart等人(1985)cell41:521-530)、sv40启动子、二氢叶酸还原酶启动子、β-肌动蛋白启动子、磷酸甘油激酶(pgk)启动子和ef1α启动子。
还由术语“调节元件”涵盖的是增强子元件,例如wpre;cmv增强剂;htlv-1的ltr中的r-u5'区段(takebe等人(1988)mol.cell.biol.8:466-472);sv40增强子;以及兔β-珠蛋白的外显子2和3之间的内含子序列(o’hare等人(1981)proc.natl.acad.sci.usa.78(3):1527-31)。本领域技术人员应了解,表达载体的设计可以取决于此类因素如待转化的宿主细胞的选择、所需表达水平等。可以将载体引入宿主细胞内,以从而产生由如本文所述的核酸编码的转录物、蛋白质或肽,包括融合蛋白或肽(例如,成簇规律间隔短回文重复(crispr)转录物、蛋白质、酶、其突变体形式、其融合蛋白等)。有利的载体包括慢病毒和腺伴随病毒,并且还可以选择此类载体的类型用于靶向特定类型的细胞。
术语“多核苷酸”、“核苷酸”、“核苷酸序列”、“核酸”和“寡核苷酸”可互换使用。它们指任何长度的聚合形式的核苷酸(脱氧核糖核苷酸或核糖核苷酸,或其类似物)。多核苷酸可以具有任何三维结构,并且可以执行已知或未知的任何功能。下述是多核苷酸的非限制性实例:基因或基因片段的编码或非编码区、由连锁分析定义的基因座(loci)(基因座(locus))、外显子、内含子、信使rna(mrna)、转移rna、核糖体rna、短干扰rna(sirna)、短发夹rna(shrna)、微rna(mirna)、核酶、cdna、重组多核苷酸、分支多核苷酸、质粒、载体、任何序列的分离的dna、任何序列的分离的rna、核酸探针和引物。多核苷酸可以包含一种或多种修饰的核苷酸,例如甲基化的核苷酸和核苷酸类似物。如果存在,则可以在聚合物组装之前或之后赋予对核苷酸结构的修饰。核苷酸序列可以被非核苷酸组分中断。聚合后可以进一步修饰多核苷酸,例如通过与标记组分缀合。
在本文公开主题的方面,术语“嵌合rna”、“嵌合指导rna”、“指导rna”、“单一指导rna”和“合成指导rna”可互换使用,并且指包含指导序列的多核苷酸序列。术语“指导序列”指指导rna内指定靶位点的约20bp的序列,并且可以与术语“指导”或“间隔物”互换使用。
如本文使用的,术语“野生型”是由本领域技术人员理解的术语,并且意指如它在自然界中出现的生物、菌株、基因或特征的典型形式,如区别于突变体或变体形式。
如本文使用的,术语“变体”应当视为意指具有偏离自然界中出现的模式的品质的展示。
术语“非天然存在的”或“改造的”可互换使用并且指示人工参与。当提及核酸分子或多肽时,该术语意指核酸分子或多肽至少基本上不含它们与之在自然界中天然结合且如自然界中发现的至少一种其他组分。
“互补性”指核酸通过传统的沃森-克里克或其他非传统类型与另一种核酸序列形成氢键的能力。百分比互补性指示核酸分子中可以与第二核酸序列形成氢键(例如,沃森-克里克碱基配对)的残基的百分比(例如,10个中的5、6、7、8、9、10个是50%、60%、70%、80%、90%和互补的)。“完全互补”意指核酸序列的所有邻接残基将与第二核酸序列中相同数目的邻接残基氢键键合。如本文使用的,“基本上互补”指在8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、30、35、40、45、50个或更多个核苷酸的区域上,其为至少60%、65%、70%、75%、80%、85%、90%、95%.97%、98%、99%或的互补性程度,或者指在严格条件下杂交的两种核酸。
如本文使用的,关于杂交的“严格条件”指在其下与靶序列具有互补性的核酸占优势地与靶序列杂交,并且基本上不与非靶序列杂交的条件。严格条件一般是序列依赖性的,并且取决于许多因素而变。一般而言,序列越长,在其下序列与其靶序列特异性杂交的温度越高。tijssen(1993),laboratorytechniquesinbiochemistryandmolecularbiology-hybridizationwithnucleicacidprobes**部分第二章"overviewofprinciplesofhybridizationandthestrategyofnucleicacidprobeassay",elsevier,n.y中详细描述了严格条件的非限制性实例。
“杂交”指其中一种或多种多核苷酸反应以形成复合物的反应,所述复合物经由核苷酸残基的碱基之间的氢键合稳定。氢键合可以通过沃森克里克碱基配对、hoogstein结合或以任何其他序列特异性方式发生。复合物可以包含形成双链体结构的两条链,形成多链复合物的三条或更多条链,单一自杂交链或这些的任何组合。杂交反应可以构成更广泛过程中的步骤,例如pcr的起始或通过酶的多核苷酸切割。能够与给定序列杂交的序列称为给定序列的“互补体”。
如本文使用的,“表达”指多核苷酸通过其从dna模板转录(为例如mrna或其他rna转录物)的过程和/或转录的mrna随后通过其翻译成肽、多肽或蛋白质的过程。转录物和编码的多肽可以统称为“基因产物”。如果多核苷酸衍生自基因组dna,则表达可以包括mrna在真核细胞中的剪接。
术语“多肽”、“肽”和“蛋白质”在本文中可互换使用,以指任何长度的氨基酸聚合物。聚合物可以是线性或分支的,它可以包含修饰的氨基酸,并且它可以被非氨基酸中断。该术语还涵盖已修饰的氨基酸聚合物;例如,二硫键形成、糖基化、脂化、乙酰化、磷酸化或任何其他操作,例如与标记组分的缀合。
如本文使用的,术语“氨基酸”包括天然和/或非天然或合成的氨基酸,包括甘氨酸和d或l光学异构体两者,以及氨基酸类似物和拟肽。
除非另有说明,否则本文公开主题的实践采用免疫学、生物化学、化学、分子生物学、微生物学、细胞生物学、基因组学和重组dna的常规技术,所述技术在本领域的技术内(sambrook,fritsch和maniatis(1989)molecularcloning:alaboratorymanual,第2版;ausubel等人,编辑(1987)currentprotocolsinmolecularbiology);macpherson等人,编辑(1995)methodsinenzymology(academicpress,inc.):pcr2:apracticalapproach);harlow和lane,编辑(1988)antibodies,alaboratorymanual;freshney,ed.(1987)animalcellculture)。
本文公开主题的几个方面涉及包含一种或多种载体的载体系统或者载体本身。载体可以设计用于在原核细胞或真核细胞中表达crispr转录物(例如核酸转录物、蛋白质或酶)。例如,crispr转录物可以在细菌细胞例如大肠杆菌(escherichiacoli)、昆虫细胞(使用杆状病毒表达载体)、酵母细胞或哺乳动物细胞中表达。合适的宿主细胞在goeddel(1990)geneexpressiontechnology:methodsinenzymology185,academicpress,sandiego,calif中进一步讨论。可替代地,重组表达载体可以在体外转录且翻译,例如使用t7启动子调节序列和t7聚合酶。
载体可以在原核生物中引入且进行倍增。在一些实施方案中,将原核生物用于扩增待引入真核细胞内的载体或作为待引入真核细胞内的载体的产生中的中间载体(例如,扩增作为病毒载体包装系统的部分的质粒)的拷贝。在一些实施方案中,原核生物用于扩增载体的拷贝并且表达一种或多种核酸,例如以提供用于递送至宿主细胞或宿主生物的一种或多种蛋白质的来源。蛋白质在原核生物中的表达更经常在大肠杆菌中用含有组成型或诱导型启动子的载体进行,所述启动子指导融合蛋白或非融合蛋白的表达。
融合载体向其中编码的蛋白质,例如重组蛋白质的氨基末端添加许多氨基酸。此类融合载体可以发挥一个或多个目的,例如:(i)增加重组蛋白的表达;(ii)增加重组蛋白的溶解度;以及(iii)通过在亲和纯化中充当配体来帮助重组蛋白的纯化。通常,在融合表达载体中,在融合部分和重组蛋白的连接处引入蛋白酶解切割位点,以致使融合蛋白纯化后重组蛋白与融合部分的分离。此类酶及其同源识别序列包括因子xa、凝血酶和肠激酶。示例性融合表达载体包括pgex(pharmaciabiotechinc;smith和johnson(1988)gene67:31-40)、pmal(newenglandbiolabs,beverly,mass.)和prit5(pharmacia,piscataway,n.j.),其将谷胱甘肽s-转移酶(gst)、麦芽糖e结合蛋白或蛋白a分别与靶重组蛋白融合。
合适的诱导型非融合大肠杆菌表达载体的实例包括ptrc(amrann等人(1988)gene69:301-315)和pet11d(studier等人(1990)geneexpressiontechnology:methodsinenzymology185,academicpress,sandiego,calif.)。
在一些实施方案中,载体是酵母表达载体。用于在酵母酿酒酵母(saccharomycescerivisae)中表达的载体的实例包括pyepsec1(baldari,等人(1987)emboj.6:229-234)、pmfa(kuijan和herskowitz(1982)cell30:933-943)、pjry88(schultz等人(1987)gene54:113-123)、pyes2(invitrogencorporation,sandiego,calif.)和picz(invitrogencorp,sandiego,calif.)。
在一些实施方案中,载体能够使用哺乳动物表达载体驱动一种或多种序列在哺乳动物细胞中的表达。哺乳动物表达载体的实例包括pcdm8(seed(1987)nature329:840)和pmt2pc(kaufman等人(1987)emboj.6:187-195)。当在哺乳动物细胞中使用时,表达载体的控制功能通常由一种或多种调节元件提供。例如,常用的启动子衍生自多瘤病毒、腺病毒2、巨细胞病毒、猿猴病毒40以及本文公开和本领域已知的其他病毒。对于用于原核细胞和真核细胞两者的其他合适的表达系统,参见例如sambrook等人(1989)molecularcloning:alaboratorymanual.第2版,coldspringharborlaboratory,coldspringharborlaboratorypress,coldspringharbor,n.y.的第16章和第17章。
在一些实施方案中,重组哺乳动物表达载体能够优先指导核酸在特定细胞类型中的表达(例如,组织特异性调节元件用于表达核酸)。组织特异性调节元件是本领域已知的。合适的组织特异性启动子的非限制性实例包括白蛋白启动子(肝特异性;pinkert等人(1987)genesdev.1:268-277),淋巴样特异性启动子(calame和eaton(1988)adv.immunol.43:235-275),特别是t细胞受体(winoto和baltimore(1989)emboj.8:729-733)和免疫球蛋白(baneiji等人(1983)cell33:729-740;queen和baltimore(1983)cell33:741-748)的启动子,神经元特异性启动子(例如,神经丝启动子;byrne和ruddle(1989)proc.natl.acad.sci.usa86:5473-5477),胰腺特异性启动子(edlund等人(1985)science230:912-916)和乳腺特异性启动子(例如,乳清启动子;美国**号4,873,316和欧洲申请公开号264,166)。还涵盖发育调节的启动子,例如鼠hox启动子(kessel和gruss(1990)science249:374-379)和甲胎蛋白启动子(campes和tilghman(1989)genesdev.3:537-546)。
在一些实施方案中,调节元件可操作地连接至crispr系统的一种或多种元件,以便驱动crispr系统的一种或多种元件的表达。一般而言,crispr(成簇规律间隔短回文重复),也称为spidr(间隔穿插直接重复(spacerintersperseddirectrepeats)),构成通常对特定细菌物种特异性的dna基因座家族。crispr基因座包含在大肠杆菌中识别的不同种类的散布短序列重复(ssr)(ishino等人(1987)j.bacteriol.,169:5429-5433;以及nakata等人(1989)j.bacteriol.,171:3553-3556)和相关基因。已在极端嗜盐古菌(haloferaxmediterranei)、化脓性链球菌、鱼腥藻属(anabaena)和结核分枝杆菌(mycobacteriumtuberculosis)中鉴定了类似的散布ssr(groenen等人(1993)mol.microbiol.,10:1057-1065;hoe等人(1999)emerg.infect.dis.,5:254-263;masepohl等人(1996)biochim.biophys.acta1307:26-30;和mojica等人(1995)mol.microbiol.,17:85-93)。crispr基因座通常与其他ssr的不同之处在于重复的结构,其已被命名为短规律间隔重复(srsr)(janssen等人(2002)omicsj.integ.biol.,6:23-33;和mojica等人(2000)mol.microbiol.,36:244-246)。一般而言,重复是以簇形式出现的短元件,所述簇由具有基本上恒定长度的独特间插序列规律地间隔开(mojica等人(2000)mol.microbiol.,36:244-246)。尽管重复序列在菌株之间是高度保守的,但散布重复的数目和间隔区的序列通常因菌株而异(vanembden等人(2000)j.bacteriol.,182:2393-2401)。已在多于40种原核生物中鉴定了crispr基因座(例如,jansen等人(2002)mol.microbiol.,43:1565-1575;和mojica等人(2005)j.mol.evol.60:174-82),所述原核生物包括但不限于气火菌属(aeropyrum)、热棒菌属(pyrobaculum)、硫化叶菌属(sulfolobus)、古生球菌属(archaeoglobus)、halocarcula、甲烷短杆菌属(methanobacteriumn)、甲烷球菌属(methanococcus)、甲烷八叠球菌属(methanosarcina)、甲烷火菌属(methanopyrus)、火球菌属(pyrococcus)、嗜酸菌属(picrophilus)、thernioplasnia、棒状杆菌属(corynebacterium)、分枝杆菌属(mycobacterium)、链霉菌属(streptomyces)、产液菌属(aquifrx)、卟啉单胞菌属(porphvromonas)、绿菌属(chlorobium)、栖热菌属(thermus)、芽孢杆菌属(bacillus)、李斯特菌属(listeria)、葡萄球菌属(staphylococcus)、梭菌属(clostridium)、高温厌氧杆菌属(thermoanaerobacter)、支原体属(mycoplasma)、梭杆菌属(fusobacterium)、azarcus、色杆菌属(chromobacterium)、奈瑟菌属(neisseria)、亚硝化单胞菌属(nitrosomonas)、脱硫弧菌属(desulfovibrio)、地杆菌属(geobacter)、myrococcus、弯曲杆菌属(campylobacter)、沃林氏菌属(wolinella)、不动杆菌属(acinetobacter)、欧文氏菌属(erwinia)、埃希氏杆菌属(escherichia)、军团菌属(legionella)、甲基球菌属(methylococcus)、巴斯德菌属(pasteurella)、发光杆菌属(photobacterium)、沙门氏菌属(salmonella)、黄单胞菌属(xanthomonas)、耶尔森氏菌属(yersinia)、密螺旋体属(treponema)和栖热袍菌属(thermotoga)。
一般而言,“crispr系统”共同指涉及crispr相关(“cas”)基因表达或指导crispr相关(“cas”)基因活性的转录物和其他元件,包括编码cas基因的序列、指导序列(在内源crispr系统的背景下也称为“间隔物”)、或来自crispr基因座的其他序列和转录物。在一些实施方案中,crispr系统的一种或多种元件衍生自i型、ii型或iii型crispr系统。在一些实施方案中,crispr系统的一种或多种元件衍生自包含内源crispr系统的特定生物,例如化脓性链球菌。一般而言,crispr系统的特征在于促进靶序列的位点处的crispr复合物形成的元件(在内源crispr系统的背景下也称为前间区序列)。
在crispr复合物形成的背景下,“靶序列”指指导序列设计为针对其具有互补性的序列,其中靶序列和指导序列之间的杂交促进crispr复合物的形成。不一定需要完全互补性,条件是存在足够的互补性以引起杂交且促进crispr复合物的形成。靶序列可以包含任何多核苷酸,例如dna或rna多核苷酸。在一些实施方案中,靶序列位于细胞的核或细胞质中。在一些实施方案中,靶序列可以在真核细胞的细胞器内,例如线粒体或叶绿体。可以用于重组到包含靶序列的靶基因座内的序列或模板称为“编辑模板”或“编辑多核苷酸”或“编辑序列”。在本文公开主题的方面,外源模板多核苷酸可以称为编辑模板。在本文公开主题的一个方面,重组是同源重组。
在一些实施方案中,载体包含一个或多个插入位点,例如限制性核酸内切酶识别序列(也称为“克隆位点”)。在一些实施方案中,一个或多个插入位点(例如,约或多于约1、2、3、4、5、6、7、8、9、10个或更多个插入位点)位于一种或多种载体的一个或多个序列元件的上游和/或下游。当使用多个不同的指导序列时,单个表达构建体可以用于将crispr活性靶向细胞内的多个不同的相应靶序列。例如,单个载体可以包含约或多于约1、2、3、4、5、6、7、8、9、10、15、20个或更多个指导序列。在一些实施方案中,可以提供约或多于约1、2、3、4、5、6、7、8、9、10个或更多个此类含有指导序列的载体,并且任选地递送至细胞。
在一些实施方案中,载体包含与编码crispr酶,例如cas蛋白的酶编码序列可操作地连接的调节元件。cas蛋白的非限制性实例包括cas1、cas1b、cas2、cas3、cas4、cas5、cas6、cas7、cas8、cas9(也称为csn1和csx12)、cas10、csy1、csy2、csy3、cse1、cse2、csc1、csc2、csa5、csn2、csm2、csm3、csm4、csm5、csm6、cmr1、cmr3、cmr4、cmr5、cmr6、csb1、csb2、csb3、csx17、csx14、csx10、csx16、csax、csx3、csx1、csx15、csf1、csf2、csf3、csf4、cpf1、c2c1、cas13a、c2c2、c2c3、其同源物或其修饰形式。这些酶是已知的;例如,化脓性链球菌cas9蛋白的氨基酸序列可以在登录号q99zw2下在swissprot数据库中找到。在一些实施方案中,未修饰的crispr酶具有dna切割活性,例如cas9。在一些实施方案中,crispr酶是cas9,并且可以是来自化脓性链球菌或肺炎链球菌的cas9。
在一些实施方案中,crispr酶指导在靶序列的位置处的一条或两条链的切割,例如在靶序列内和/或在靶序列的互补体内。在一些实施方案中,crispr酶指导距离靶序列的**个或更后一个核苷酸的约1、2、3、4、5、6、7、8、9、10、15、20、25、50、100、200、500个或更多个碱基对内的一条链或两条链的切割。在一些实施方案中,载体编码关于相应的野生型酶突变的crispr酶,使得突变的crispr酶缺乏切割含有靶序列的靶多核苷酸的一条链或两条链的能力。
在一些实施方案中,编码crispr酶的酶编码序列进行密码子优化,用于在特定细胞例如真核细胞中表达。真核细胞可以是特定生物的那些或衍生自特定生物,例如哺乳动物,包括但不限于人、小鼠、大鼠、兔、犬或非人灵长类动物。一般而言,密码子优化指通过用在该宿主细胞的基因中更频繁或更频繁使用的密码子替换天然序列的至少一个密码子(例如,约或多于约1、2、3、4、5、10、15、20、25、50个或更多个密码子),同时维持天然氨基酸序列,来修饰核酸序列用于目的宿主细胞中的增强表达的过程。各种物种显示出对于特定氨基酸的某些密码子的特别偏倚。密码子偏倚(生物之间的密码子使用中的差异)经常与信使rna(mrna)的翻译效率相关联,所述信使rna(mrna)的翻译效率依次又被认为尤其取决于待翻译的密码子的特性和特定转移rna(trna)分子的可用性。所选择的trna在细胞中的优势一般是肽合成中更频繁使用的密码子的反映。相应地,可以基于密码子优化来定制基因,用于在给定生物中的更佳基因表达。密码子使用表例如在“密码子使用数据库”中可容易获得,并且这些表可以以多种方式进行调整。参见nakamura等人(2000)nucl.acidsres.28:292。用于密码子优化特定序列用于在特定宿主细胞中表达的计算机算法也是可用的,例如geneforge(aptagen;jacobus,pa.)也是可用的。在一些实施方案中,编码crispr酶的序列中的一个或多个密码子(例如,1、2、3、4、5、10、15、20、25、50个或更多个或所有密码子)对应于关于特定氨基酸的更频繁使用的密码子。
一般而言,指导序列是与靶多核苷酸序列具有足够互补性以与靶序列杂交并且指导crispr复合物与靶序列的直接序列特异性结合的任何多核苷酸序列。在一些实施方案中,当使用合适的比对算法更佳比对时,指导序列与其相应的靶序列之间的互补性程度为约或多于约50%、60%、75%、80%、85%、90%、95%、97.5%、99%或更多。可以使用用于比对序列的任何合适的算法来确定更佳比对,所述算法的非限制性实例包括smith-waterman算法、needleman-wunsch算法、基于burrows-wheeler变换的算法(例如burrowswheeleraligner)、clustalw、clustalx、blat、novoalign(novocrafttechnologies、eland(illumina,sandiego,calif.)、soap(可在soap.genomics.org.cn处获得)和maq(可在maq.sourceforge.net处获得)。在一些实施方案中,指导序列为长度约或多于约5、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、35、40、45、50、75个或更多个核苷酸。在一些实施方案中,指导序列为长度小于约75、50、45、40、35、30、25、20、15、12个或更少的核苷酸。
可以通过任何合适的测定来评价指导序列指导crispr复合物与靶序列的序列特异性结合的能力。例如,足以形成crispr复合物的crispr系统的组件,包括待测试的指导序列,可以提供给具有相应靶序列的宿主细胞,例如通过用编码crispr序列的组件的载体转染,随后为靶序列内的优先切割的评价,例如通过如本文所述的surveyor测定。类似地,通过提供靶序列,crispr复合物的组件,包括待测试的指导序列和不同于测试指导序列的对照指导序列,并且比较在测试和对照指导序列反应之间的靶序列处的结合或切割速率,可以在试管中评估靶多核苷酸序列的切割。其他测定是可能的,并且将是本领域技术人员想到的。
可以选择指导序列以靶向任何靶序列。在一些实施方案中,靶序列是细胞基因组内的序列。示例性靶序列包括靶基因组中独特的靶序列。例如,在一些实施方案中,lca的靶序列选自seqidno:1、2、3、4、5和6;或其组合。seqidno:1、2、3、4、5和6;或其组合可以导致~1kb缺失,从cep290中去除隐藏外显子d。将seqidno:1、2、3、4、5和6与修饰形式的cas9(例如d10a切口酶)组合可以提供安全且有效的治疗方法。具有四个grna的示例性sacas9构建体在seqidno:110中示出。lca的另外的靶序列可以选自seqidno:7-109中所示的核苷酸序列。在一些实施方案中,adrp的靶序列选自seqidno:111-126或其组合。在一些实施方案中,adrp的grna序列选自seqidno:127-142或其组合。在一些实施方案中,青光眼的突变靶包括但不限于optn、tbk1、tmco1、pmm2、gmds、gas7、fndc3b、txnrd2、atxn2、cav1/cav2、p16ink4a、six6、abca1、afap1和cdkn2b-as。在一些实施方案中,青光眼的靶序列选自seqidno:143-163或其组合。在用于治疗青光眼的方法的一些实施方案中,非天然存在的crispr系统包含在polii方向上表达mcherry-组蛋白2b融合物的h1启动子与例如针对dlk、lzk的至少一种grna、或使用此处提供的筛选方法鉴定rgc存活途径的其他上游或下游组分的组合。
在一些实施方案中,靶序列可以与seqidno:1-738或788-1397中所示的核苷酸序列60%、65%、70%、75%、80%、81%、82%、83%、84%、85%、86%、87%、88%、89%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%、99.1%、99.2%、99.3%、99.4%、99.5%、99.6%、99.7%、99.8%同源。
术语“同源的”指“%同源性”并且在本文中可与术语“%同一性”互换使用,并且涉及当使用序列比对程序比对时核酸序列同一性的水平。
例如,如本文使用的,80%同源性意指与通过限定算法确定的与80%序列同一性相同的事物,并且相应地,给定序列的同源物在给定序列的长度上具有大于80%的序列同一性。序列同一性的示例性水平包括但不限于与seqidno:1-1400中所示的核苷酸序列约80%、81%、82%、83%、84%、85%、86%、87%、88%、89%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99.1%、99.2%、99.3%、99.4%、99.5%、99.6%、99.7%、99.8%或更高的序列同一性。
在一些实施方案中,crispr酶是包含一个或多个异源蛋白结构域(例如,除crispr酶之外,约或多于约1、2、3、4、5、6、7、8、9、10个或更多个结构域)的融合蛋白的部分。crispr酶融合蛋白可以包含任何另外的蛋白质序列,以及任选地在任何两个结构域之间的接头序列。可以与crispr酶融合的蛋白质结构域的实例包括但不限于表位标签、报道基因序列和具有下述活性中的一种或多种的蛋白质结构域:甲基化酶活性、去甲基化酶活性、转录激活活性、转录阻遏活性、转录释放因子活性、组蛋白修饰活性、rna切割活性和核酸结合活性。表位标签的非限制性实例包括组氨酸(his)标签、v5标签、flag标签、流感血凝素(ha)标签、myc标签、vsv-g标签和硫氧还蛋白(trx)标签。报道基因的实例包括但不限于谷胱甘肽-5-转移酶(gst)、辣根过氧化物酶(hrp)、氯霉素乙酰转移酶(cat)、β-半乳糖苷酶、β-葡糖醛酸酶、荧光素酶、绿色荧光蛋白(gfp)、hcred、dsred、青色荧光蛋白(cfp)、黄色荧光蛋白(yfp)和自体荧光蛋白包括蓝色荧光蛋白(bfp)。crispr酶可以与编码蛋白质或蛋白质片段的基因序列融合,所述蛋白质或蛋白质片段结合dna分子或结合其他细胞分子,包括但不限于麦芽糖结合蛋白(mbp)、s标签、lexadna结合结构域(dbd)融合物、gal4adna结合域融合物和单纯疱疹病毒(hsv)bp16蛋白融合物。可以形成包含crispr酶的融合蛋白的部分的另外结构域在us20110059502中描述,所述**通过引用并入本文。在一些实施方案中,加上标签的crispr酶用于鉴定靶序列的位置。
在本文公开主题的一个方面,包括但不限于谷胱甘肽-5-转移酶(gst)、辣根过氧化物酶(hrp)、氯霉素乙酰转移酶(cat)、β-半乳糖苷酶、β-葡糖醛酸酶、荧光素酶、绿色荧光蛋白(gfp)、hcred、dsred、青色荧光蛋白(cfp)、黄色荧光蛋白(yfp)和自体荧光蛋白包括蓝色荧光蛋白(bfp)的报道基因可以引入细胞内,以编码充当通过其测量基因产物表达的改变或修饰的标记物的基因产物。在本文公开主题的一个进一步实施方案中,可以经由载体将编码基因产物的dna分子引入细胞内。在本文公开主题的一个优选实施方案中,基因产物是荧光素酶。在本文公开主题的一个进一步实施方案中,基因产物的表达降低。
一般地,本文公开主题的启动子实施方案包括:1)完整的poliii启动子,其包括tata盒、近端序列元件(pse)和远端序列元件(dse);以及2)第二基础poliii启动子,其包括以相反方向与dse的5'末端融合的pse和tata盒。因其核苷酸序列命名的tata盒是poliii特异性的主要决定因素。它通常位于相对于转录序列nt.?23和?30之间的位置处,并且是转录序列开始的主要决定因素。pse通常位于nt.?45和?66之间。dse增强了基础poliii启动子的活性。在h1启动子中,pse和dse之间不存在间隙。
双向启动子由以下组成:1)完整的、常规、单向poliii启动子,其含有3个外部控制元件:dse、pse和tata盒;以及2)第二基础poliii启动子,其包括以相反方向与dse的5'末端融合的pse和tata盒。被tata结合蛋白识别的tata盒对于将poliii募集到启动子区是必需的。tata结合蛋白与tata盒的结合通过snapc与pse的相互作用得到稳定。这些元素一起正确定位poliii,使得它可以转录表达的序列。dse对于poliii启动子的完全活性也是必需的(murphy等人(1992)mol.cellbiol.12:3247-3261;mittal等人(1996)mol.cellbiol.16:1955-1965;ford和hernandez(1997)j.biol.chem.,272:16048-16055;ford等人(1998)genes,dev.,12:3528-3540;hovde等人(2002)genesdev.16:2772-2777)。通过转录因子oct-1和/或sbf/staf与其在dse内的基序的相互作用,转录增强高达100倍(kunkel和hixon(1998)nucl.acidres.,26:1536-1543)。因为正向和反向取向的基础启动子指导序列在双链dna模板的相反链上的转录,所以反向取向的基础启动子的正链附加到dse负链的5'末端。在h1启动子的控制下表达的转录物由4或5个t的连续序列终止。
在h1启动子中,dse与pse和tata盒相邻(myslinski等人(2001)nucl.acidres.29:2502-2509)。为了使序列重复更小化,通过产生杂合启动子,致使该启动子双向,其中通过附加衍生自u6启动子的pse和tata盒来控制反向中的转录。为了促进双向h1启动子的构建,还可以在反向取向的基础启动子和dse之间插入小间隔区序列。
在一些实施方案中,双向启动子包括但不限于h1、rpph1-parp2(人)、srp-rps29、7sk1-gsta4、snar-g-1-cgb1、snar-cgb2、rmrp-ccdc107、trna(lys)-aloxe3、rnu6-9-med16:trna(gly)-dpp9、rnu6-2-them259或snord13-c8orf41。
在一些实施方案中,h1启动子包含seqidno:787中所示的核苷酸序列。
在一些实施方案中,直向同源的双向启动子包括但不限于rpph1-parp2(小鼠)或rpph1-parp2(大鼠),或衍生自以下的那些:大熊猫、普通牛、普通狨、家犬、豚鼠、绿猴、霍氏树懒、九带犰狳、奥氏更格卢鼠、家马、普通刺猬、家猫、大猩猩、智人、十三纹地松鼠、非洲象、恒河猴、小家鼠、雪貂、小棕蝠、白颊长臂猿、北美鼠兔、穴兔、小耳大婴猴、绵羊、黑猩猩、东非狒狒、苏门答腊猩猩、岩石蹄兔、大狐蝠、褐家鼠、野猪、眼镜猴、树鼩、宽吻海豚、羊驼。
表7:直向同源的h1序列的实例
小家鼠
褐家鼠
奥氏更格卢鼠
十三纹地松鼠
豚鼠
北美鼠兔
(seqidno:757)
穴兔
普通狨
绿猴
恒河猴
东非狒狒
大猩猩
智人
黑猩猩
苏门答腊猩猩
白颊长臂猿
眼镜猴
小耳大婴猴
树鼩
大熊猫
雪貂
家犬
家猫
家马
小棕蝠
大狐蝠
普通牛
绵羊
宽吻海豚
羊驼
野猪
普通刺猬
霍氏树懒
九带犰狳
非洲象
岩石蹄兔
b.方法
在一些实施方案中,本文公开主题还提供了改变真核细胞中的一种或多种基因产物表达的方法,其中所述细胞包含编码一种或多种基因产物的dna分子,所述方法包括向所述细胞内引入先前在wo2015/195621(通过引用整体并入本文)中描述的修饰的非天然存在的crispr系统。此类修饰使用某些grna,其靶向视网膜变性相关基因,例如但不限于lca10cep290基因、视紫红质、双亮氨酸拉链激酶(dlk)、亮氨酸拉链激酶(lzk)、jnk1-3、mkk4、mkk7、atf2、jun、mef2a、sox11或puma。在一些实施方案中,该方法包括向细胞中引入组合物,所述组合物包含(a)包含一种或多种载体的非天然存在的核酸酶系统(例如,crispr),所述载体包含:i)与编码核酸酶系统指导rna(grna)的至少一种核苷酸序列可操作地连接的启动子(例如,双向h1启动子),其中所述grna与主体的细胞中的dna分子的靶序列杂交,并且其中所述dna分子编码在细胞中表达的一种或多种基因产物;以及ii)在细胞中可操作的调节元件,其与编码基因组靶向核酸酶(例如cas9蛋白)的核苷酸序列可操作地连接,其中组件(i)和(ii)位于系统的相同或不同载体上,其中所述grna靶向所述靶序列且与之杂交,并且核酸酶切割dna分子以改变一种或多种基因产物的表达。在一些实施方案中,将系统包装到单个腺伴随病毒(aav)颗粒内。在一些实施方案中,腺伴随病毒(aav)可以包含51种人腺病毒血清型中的任一种(例如,血清型2、5或35)。在一些实施方案中,该系统使一种或多种基因产物失活。在一些实施方案中,核酸酶系统切除至少一个基因突变。在一些实施方案中,启动子包含:a)提供在编码grna的至少一种核苷酸序列的一个方向上的转录的控制元件;以及b)提供在编码基因组靶向核酸酶的核苷酸序列的相反方向上的转录的控制元件。在一些实施方案中,cas9蛋白进行密码子优化用于在细胞中表达。在一些实施方案中,启动子与至少一个、两个、三个、四个、五个、六个、七个、八个、九个或十个grna可操作地连接。在一些实施方案中,靶序列是cep290基因(例如,lca10cep290基因)中的突变。在一些实施方案中,关于cep290的靶序列选自seqidno:1-109、164-356、735-738中所示的核苷酸序列,或其组合。在一些实施方案中,靶序列包含可操作地连接的seqidno:1、2、3和4。在一些实施方案中,载体包含seqidno:110中所示的核苷酸序列。在一些实施方案中,一种或多种基因产物是视紫红质。在一些实施方案中,靶序列是视紫红质基因中的突变。在一些实施方案中,靶序列是在视紫红质基因的r135处的突变(例如,r135g、r135w、r135l)。在一些实施方案中,关于视紫红质r135的靶序列选自seqidno:111-126中所示的核苷酸序列、或其组合。在一些实施方案中,关于视紫红质r135的grna序列选自seqidno:127-142中所示的核苷酸序列、或其组合。在一些实施方案中,一种或多种基因产物是双亮氨酸拉链激酶(dlk)、亮氨酸拉链激酶(lzk)、jnk1-3、mkk4、mkk7、atf2、jun、mef2a、sox11或puma或其组合。在一些实施方案中,青光眼的突变靶包括但不限于optn、tbk1、tmco1、pmm2、gmds、gas7、fndc3b、txnrd2、atxn2、cav1/cav2、p16ink4a、six6、abca1、afap1和cdkn2b-as。在一些实施方案中,关于青光眼的靶序列选自seqidno:143-163中所示的核苷酸序列、或其组合。
在一些实施方案中,本文公开主题还提供了改变细胞中的一种或多种基因产物表达的方法,其中所述细胞包含编码一种或多种基因产物的dna分子,所述方法包括向所述细胞内引入非天然存在的crispr系统,所述crispr系统包含一种或多种载体,所述载体包含:a)与编码crispr系统指导rna(grna)的至少一种核苷酸序列可操作地连接的h1启动子,其中所述grna与dna分子的靶序列杂交;以及b)在细胞中可操作的调节元件,其与编码cas9蛋白的核苷酸序列可操作地连接,其中组件(a)和(b)位于系统的相同或不同载体上,其中所述grna靶向所述靶序列且与之杂交,并且cas9蛋白切割dna分子以改变一种或多种基因产物的表达。
在一些实施方案中,本文公开主题还提供了改变真核细胞中的一种或多种基因产物表达的方法,其中所述细胞包含编码一种或多种基因产物的dna分子,所述方法包括向所述细胞内引入非天然存在的crispr系统,所述crispr系统包含一种或多种载体,所述载体包含:a)与编码crispr系统指导rna(grna)的至少一种核苷酸序列可操作地连接的h1启动子,其中所述grna与dna分子的靶序列杂交;以及b)在真核细胞中可操作的调节元件,其与编码ii型cas9蛋白的核苷酸序列可操作地连接,其中组件(a)和(b)位于系统的相同或不同载体上,由此所述grna靶向所述靶序列且与之杂交,并且cas9蛋白切割dna分子,并且由此改变一种或多种基因产物的表达。在一个方面,靶序列可以是以任何核苷酸开始的靶序列,例如n20ngg。在一些实施方案中,靶序列包含核苷酸序列an19ngg。在一些实施方案中,靶序列包含核苷酸序列gn19ngg。在一些实施方案中,靶序列包含核苷酸序列cn19ngg。在一些实施方案中,靶序列包含核苷酸序列tn19ngg。在一些实施方案中,靶序列包含核苷酸序列an19ngg或gn19ngg。在另一个方面,cas9蛋白进行密码子优化用于在细胞中表达。在另外一个方面,cas9蛋白进行密码子优化用于在真核细胞中表达。在一个进一步方面,真核细胞是哺乳动物细胞或人细胞。在另一个方面,一种或多种基因产物的表达降低。
本文公开主题还提供了改变真核细胞中的一种或多种基因产物表达的方法,其中所述细胞包含编码一种或多种基因产物的dna分子,所述方法包括向所述细胞内引入非天然存在的crispr系统,所述crispr系统包含含有双向h1启动子的载体,其中所述双向h1启动子包含:a)提供在编码crispr系统指导rna(grna)的至少一种核苷酸序列的一个方向上的转录的控制元件,其中所述grna与dna分子的靶序列杂交;以及b)提供在编码ii型cas9蛋白的核苷酸序列的相反方向上的转录的控制元件,由此所述grna靶向所述靶序列且与之杂交,并且cas9蛋白切割dna分子,并且由此改变一种或多种基因产物的表达。在一个方面,靶序列可以是以任何核苷酸开始的靶序列,例如n20ngg。在一些实施方案中,靶序列包含核苷酸序列an19ngg。在一些实施方案中,靶序列包含核苷酸序列gn19ngg。在一些实施方案中,靶序列包含核苷酸序列cn19ngg。在一些实施方案中,靶序列包含核苷酸序列tn19ngg。在另一个方面,靶序列包含核苷酸序列an19ngg或gn19ngg。在另外一个方面,cas9蛋白进行密码子优化用于在细胞中表达。在另一个方面,cas9蛋白进行密码子优化用于在真核细胞中表达。在一个进一步方面,真核细胞是哺乳动物细胞或人细胞。在另一个方面,一种或多种基因产物的表达降低。
在一些方面,本文公开主题提供了包括将一种或多种多核苷酸,例如如本文所述的一种或多种载体、其一种或多种转录物、和/或由其转录的一种或多种蛋白质递送至宿主细胞的方法。在一些方面,本文公开主题进一步提供了通过此类方法产生的细胞,以及包含此类细胞或由此类细胞产生的生物(例如动物、植物或真菌)。在一些实施方案中,将与指导序列组合(并且任选地与之复合)的crispr酶递送至细胞。常规的基于病毒和非病毒的基因转移方法可以用于在哺乳动物细胞或靶组织中引入核酸。此类方法可以用于将编码crispr系统组件的核酸施用于培养中的细胞或宿主生物中的细胞。非病毒载体递送系统包括dna质粒、rna(例如本文所述载体的转录物)、裸露核酸和与递送媒介物复合的核酸,例如脂质体。病毒载体递送系统包括dna和rna病毒,其在递送至细胞后具有游离型或整合的基因组。关于基因治疗程序的综述,参见anderson(1992)science256:808-813;nabel和felgner(1993)tibtech11:211-217;mitani和caskey(1993)tibtech11:162-166;dillon(1993)tibtech11:167-175;miller(1992)nature357:455-460;vanbrunt(1998)biotechnology6(10):1149-1154;vigne(1995)restorativeneurologyandneuroscience8:35-36;kremer和perricaudet(1995)britishmedicalbulletin51(1):31-44;haddada等人(1995)currenttopicsinmicrobiologyandimmunology.doerfler和bohm(编辑);以及yu等人(1994)genetherapy1:13-26。
核酸的非病毒递送方法包括脂质转染、核转染、显微注射、基因枪、病毒体、脂质体、免疫脂质体、聚阳离子或脂质:核酸缀合物、裸露dna、人工病毒粒子和试剂增强的dna摄取。脂转染在例如美国**号5,049,386、4,946,787;和4,897,355)中描述;并且脂转染试剂是商业销售的(例如transfectam?和lipofectin?)。适合于多核苷酸的有效受体识别脂转染的阳离子和中性脂质包括felgner,wo91/17424;wo91/16024的那些。递送可以针对细胞(例如体外或离体施用)或靶组织(例如体内施用)。
脂质:核酸复合物,包括靶向脂质体,例如免疫脂质复合物的制备,是本领域技术人员众所周知的(例如,crystal(1995)science270:404-410;blaese等人(1995)cancergenether.2:291-297:behr等人(1994)bioconjugatechem.5:382-389;remy等人(1994)bioconjugatechem.5:647-654;gao等人(1995)genetherapy2:710-722;ahmad等人(1992)cancerres.52:4817-4820;美国**号4,186,183、4,217,344、4,235,871、4,261,975、4,485,054、4,501,728、4,774,085、4,837,028和4,946,787)。
基于rna或dna病毒的系统用于递送核酸的用途利用高度进化的方法,所述方法用于将病毒靶向体内的特定细胞并且将病毒载量运输至核。病毒载体可以直接施用于患者(体内),或者它们可以用于在体外处理细胞,并且可以任选地将修饰的细胞施用于患者(离体)。常规的基于病毒的系统可以包括用于基因转移的逆转录病毒、慢病毒、腺病毒、腺伴随和单纯疱疹病毒载体。在宿主基因组中的整合对于逆转录病毒、慢病毒和腺伴随病毒基因转移方法是可能的,经常导致插入的转基因的长期表达。另外,已在许多不同细胞类型和靶组织中观察到高转导效率。
逆转录病毒的向性可以通过掺入外来包膜蛋白,扩展靶细胞的潜在靶群体来改变。慢病毒载体是逆转录病毒载体,其能够转导或感染非分裂细胞并且通常产生高病毒滴度。逆转录病毒基因转移系统的选择因此将取决于靶组织。逆转录病毒载体由顺式作用长末端重复组成,具有高达6-10kb外来序列的包装能力。更小顺式作用ltr足以用于载体的复制和包装,所述载体随后用于将治疗基因整合到靶细胞内,以提供**的转基因表达。广泛使用的逆转录病毒载体包括基于鼠白血病病毒(mulv)、长臂猿白血病病毒(galv)、猴免疫缺陷病毒(siv)、人免疫缺陷病毒(hiv)及其组合的那些载体(例如,buchscher等人(1992)j.virol.66:2731-2739;johann等人(1992)j.virol.66:1635-1640;sommnerfelt等人(1990)j.virol.176:58-59;wilson等人(1989)j.virol.63:2374-2378;miller等人(1991)j.virol.65:2220-2224;pct/us94/05700)。在其中优选瞬时表达的应用中,可以使用基于腺病毒的系统。基于腺病毒的载体在许多细胞类型中能具有非常高的转导效率,并且不需要细胞分裂。使用此类载体,已获得了高滴度和水平的表达。该载体可以在相对简单的系统中大量生产。腺伴随病毒(“aav”)载体也可以用于用靶核酸转导细胞,例如,在核酸和肽的体外生产中,以及用于体内和离体基因治疗程序(例如,west等人(1987)virology160:38-47;美国**号4,797,368;wo93/24641;kotin(1994)humangenetherapy5:793-801;muzyczka(1994)j.clin.invest.94:1351。重组aav载体的构建在许多出版物中描述,包括美国**号5,173,414;tratschin等人(1985)mol.cell.biol.5:3251-3260;tratschin等人(1984)mol.cell.biol.4:2072-2081;hermonat和muzyczka(1984)proc.natl.acad.sci.u.s.a.81:6466-6470;以及samulski等人(1989)j.virol.63:03822-3828。
包装细胞通常用于形成能够感染宿主细胞的病毒颗粒。此类细胞包括包装腺病毒的293细胞和包装逆转录病毒的ψ2细胞或pa317细胞。用于基因治疗中的病毒载体通常通过产生将核酸载体包装到病毒颗粒内的细胞系来生成。载体通常含有包装和随后整合到宿主内所需的更小病毒序列,其他病毒序列被用于表达多核苷酸的表达盒取代。缺失的病毒功能通常由包装细胞系反式提供。例如,用于基因治疗中的aav载体通常仅具有来自aav基因组的itr序列,其是包装和整合到宿主基因组内所必需的。病毒dna包装在细胞系中,所述细胞系含有编码其他aav基因的辅助质粒,即rep和cap,但缺乏itr序列。细胞系也可以用腺病毒作为辅助物进行感染。辅助病毒促进来自辅助质粒的aav载体的复制和aav基因的表达。由于缺乏itr序列,辅助质粒未以显著量包装。由腺病毒的污染可以通过例如腺病毒对其比aav更敏感的热处理来减少。用于将核酸递送至细胞的另外方法是本领域技术人员已知的。参见例如,us20030087817,其通过引用并入本文。
在一些实施方案中,用本文所述的一种或多种载体瞬时或非瞬时转染宿主细胞。在一些实施方案中,细胞在其天然存在于主体中时被转染。在一些实施方案中,转染的细胞取自主体。在一些实施方案中,细胞衍生自取自主体的细胞,例如细胞系。用于组织培养的广泛多样的细胞系是本领域已知的。细胞系的实例包括但不限于c8161、ccrf-cem、molt、mimcd-3、nhdf、hela-s3、huh1、huh4、huh7、huvec、hasmc、hekn、heka、miapacell、panel、pc-3、tf1、ctll-2、c1r、rat6、cv1、rpte、a10、t24、j82、a375、arh-77、calu1、sw480、sw620、skov3、sk-ut、caco2、p388d1、sem-k2、wehi-231、hb56、tib55、jurkat、j45.01、lrmb、bcl-1、bc-3、ic21、dld2、raw264.7、nrk、nrk-52e、mrc5、mef、hepg2、helab、helat4、cos、cos-1、cos-6、cos-m6a、bs-c-1猴肾上皮、balb/3t3小鼠胚胎成纤维细胞、3t3swiss、3t3-l1、132-d5人胎儿成纤维细胞;10.1小鼠成纤维细胞、293-t、3t3、721、9l、a2780、a2780adr、a2780cis、a172、a20、a253、a431、a-549、alc、b16、b35、bcp-1细胞、beas-2b、bend.3、bhk-21、br293、bxpc3、c3h-10t1/2、c6/36、cal-27、cho、cho-7、cho-ir、cho-k1、cho-k2、cho-t、chodhfr?/?、cor-l23、cor-l23/cpr、cor-l23/5010、cor-l23/r23、cos-7、cov-434、cmlt1、cmt、ct26、d17、dh82、du145、ducap、el4、em2、em3、emt6/ar1、emt6/ar10.0、fm3、h1299、h69、hb54、hb55、hca2、hek-293、hela、hepa1c1c7、hl-60、hmec、ht-29、jurkat、jy细胞、k562细胞、ku812、kcl22、kg1、kyo1、lncap、ma-mei1-48、mc-38、mcf-7、mcf-10a、mda-mb-231、mda-mb-468、mda-mb-435、mdckii、mdckii、mor/0.2r、mono-mac6、mtd-1a、myend、nci-h69/cpr、nci-h69/lx10、nci-h69/lx20、nci-h69/lx4、nih-3t3、nalm-1、nw-145、opcn/opct细胞系、peer、pnt-1a/pnt2、renca、rin-5f、rma/rmas、saos-2细胞、sf-9、skbr3、t2、t-47d、t84、thp1细胞系、u373、u87、u937、vcap、vero细胞、wm39、wt-49、x63、yac-1、yar及其转基因变种。细胞系可从本领域技术人员已知的各种来源获得(参见例如,美国典型培养物保藏中心(atcc)(manassus,va.))。在一些实施方案中,用本文所述的一种或多种载体转染的细胞用于建立包含一种或多种载体衍生序列的新细胞系。在一些实施方案中,用如本文所述的crispr系统的组件瞬时转染(例如通过一种或多种载体的瞬时转染,或用rna转染),并且通过crispr复合物的活性修饰的细胞,用于建立包含含有修饰但缺乏任何其他外源序列的细胞的新细胞系。在一些实施方案中,用本文所述的一种或多种载体瞬时或非瞬时转染的细胞,或衍生自此类细胞的细胞系用于评价一种或多种测试化合物。
在一些实施方案中,本文描述的一种或多种载体用于产生非人转基因动物。在一些实施方案中,转基因动物是哺乳动物,例如小鼠、大鼠或兔。在某些实施方案中,生物或主体是植物。用于产生转基因动物的方法是本领域已知的,并且一般以例如本文所述的细胞转染方法开始。
在一个方面,本文公开主题提供了修饰真核细胞中的靶多核苷酸的方法,所述方法可以是体内、离体或体外。在一些实施方案中,该方法包括从人或非人动物取样细胞或细胞群体,并且修饰一种或多种细胞。培养可以在离体的任何阶段发生。甚至可以将一种或多种细胞重新引入非人动物内。
在一个方面,本文公开主题提供了修饰真核细胞中的靶多核苷酸的方法。在一些实施方案中,该方法包括允许crispr复合物与靶多核苷酸结合,以实现靶多核苷酸的切割,从而修饰靶多核苷酸,其中所述crispr复合物包含与指导序列复合的crispr酶,所述指导序列与靶多核苷酸内的靶序列杂交。
在一个方面,本文公开主题提供了修饰真核细胞中的多核苷酸表达的方法。在一些实施方案中,该方法包括允许crispr复合物与多核苷酸结合,使得结合导致多核苷酸的表达增加或降低;其中所述crispr复合物包含与指导序列复合的crispr酶,所述指导序列与多核苷酸内的靶序列杂交。
在一个方面,本文公开主题提供了关于使用crispr系统的一种或多种元件的方法。本文公开主题的crispr复合物提供了用于修饰靶多核苷酸的有效手段。本文公开主题的crispr复合物具有广泛多样的用途,包括在多种细胞类型中修饰(例如,缺失、插入、易位、失活、激活)靶多核苷酸。像这样,本文公开主题的crispr复合物在例如基因治疗、药物筛选、疾病诊断和预后中具有广谱应用。示例性的crispr复合物包含与指导序列复合的crispr酶,所述指导序列与靶多核苷酸内的靶序列杂交。
crispr复合物的靶多核苷酸可以是对于真核细胞内源或外源的任何多核苷酸。例如,靶多核苷酸可以是位于真核细胞的细胞核中的多核苷酸。靶多核苷酸可以是编码基因产物(例如蛋白质)的序列或非编码序列(例如调节多核苷酸或无用dna)。不希望受理论束缚,认为靶序列应与pam(前间区序列邻近基序)即由crispr复合物识别的短序列结合。pam的**序列和长度要求根据所用的crispr酶而不同,但pam通常是与前间区序列(即,靶序列)相邻的2-5个碱基对序列。pam序列的实例在下文实施例节段中给出,并且技术人员将能够鉴定用于与给定crispr酶一起使用的进一步pam序列。
靶多核苷酸的实例包括与信号传导生化途径相关的序列,例如信号传导生化途径相关基因或多核苷酸。靶多核苷酸的实例包括疾病相关基因或多核苷酸。“疾病相关”基因或多核苷酸指任何基因或多核苷酸,与非疾病对照的组织或细胞相比,其在衍生自患病组织的细胞中以异常水平或异常形式产生转录或翻译产物。它可以是变得以异常高水平表达的基因;它可以是变得以异常低水平表达的基因,其中改变的表达与疾病的发生和/或进展相关。疾病相关基因还指具有突变或遗传变异的基因,其直接负责疾病的病因学或与负责疾病的病因学的基因处于连锁不平衡。转录或翻译的产物可以是已知的或未知的,并且可以处于正常或异常水平。
本文公开主题的实施方案还涉及与敲除基因、扩增基因和修复与视网膜病症相关的特定突变相关的方法和组合物(robertd.wells,tetsuoashizawa,geneticinstabilitiesandneurologicaldiseases,第二版academicpress,oct.13,2011-medical)。已发现串联重复序列的特定方面负责多于二十种人疾病(mcivor等人(2010)rnabiol.7(5):551-8)。可以利用crispr系统来校正基因组不稳定性的这些缺陷。
在本文公开主题的另外一个方面,crispr系统可以用于校正视网膜缺陷,其起因于由traboulsi编辑(2012)geneticdiseasesoftheeye,第二版,oxforduniversitypress中进一步描述的几种基因突变。
ii.用于治疗视网膜变性的方法
本文公开主题还提供了用于治疗视网膜变性,例如lca、adrp或青光眼的方法。在一些实施方案中,本文公开主题提供了用于治疗有此需要的主体(例如人)中的视网膜变性的方法。该方法包括以下步骤:(a)提供包含一种或多种载体的非天然存在的核酸酶系统(例如,crispr),所述载体包含:i)与编码核酸酶系统指导rna(grna)的至少一种核苷酸序列可操作地连接的启动子(例如,双向h1启动子),其中所述grna与主体的细胞(例如,视网膜光感受器或神经节细胞)中的dna分子的靶序列杂交,并且其中所述dna分子编码在细胞中表达的一种或多种基因产物;以及ii)在细胞中可操作的调节元件,其与编码基因组靶向核酸酶(例如cas9)的核苷酸序列可操作地连接,其中组件(i)和(ii)位于系统的相同或不同载体上,其中所述grna靶向所述靶序列且与之杂交,并且核酸酶切割dna分子以改变一种或多种基因产物的表达或者使一种或多种基因产物失活;以及(b)向主体的视网膜区域施用治疗有效量的系统。在一些实施方案中,将系统包装到单个腺伴随病毒(aav)颗粒(例如,aav、aav2、aav9等等)内。在一些实施方案中,核酸酶系统切除至少一个基因突变。在一些实施方案中,h1启动子包含a)提供在编码grna的至少一种核苷酸序列的一个方向上的转录的控制元件;以及b)提供在编码基因组靶向核酸酶的核苷酸序列的相反方向上的转录的控制元件。在一些实施方案中,启动子与至少一个、两个、三个、四个、五个、六个、七个、八个、九个或十个grna可操作地连接。在一些实施方案中,视网膜变性选自lca1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17和18。在一些实施方案中,视网膜变性是lca10。在治疗lca的一些实施方案中,靶序列选自lca10cep290基因。在治疗lca的一些实施方案中,靶序列位于lca10cep290基因中,并且选自seqidno:1-109、164-356、735-738中所示的核苷酸序列,或其组合(例如,可操作地连接的seqidno:1、2、3和4)。在治疗lca的一些实施方案中,载体包含seqidno:110中所示的核苷酸序列。在一些实施方案中,视网膜变性是adrp。在治疗adrp的一些实施方案中,靶序列是视紫红质基因中的突变。在治疗adrp的一些实施方案中,靶序列是在视紫红质基因的r135处的突变。在一些实施方案中,在r135处的突变选自r135g、r135w、r135l。在治疗adrp的一些实施方案中,靶序列选自seqidno:111-126中所示的核苷酸序列、或其组合。在治疗adrp的一些实施方案中,grna序列选自seqidno:127-142中所示的核苷酸序列、或其组合。在一些实施方案中,视网膜变性是青光眼。在治疗青光眼的一些实施方案中,一种或多种基因产物是双亮氨酸拉链激酶(dlk)、亮氨酸拉链激酶(lzk)、jnk1-3、mkk4和mkk7、atf2、jun、mef2a、sox11或puma、或其组合。在治疗青光眼的一些实施方案中,使用下文实施例4中描述的基于rna的筛选鉴定一种或多种基因产物。在一些实施方案中,青光眼的突变靶包括但不限于optn、tbk1、tmco1、pmm2、gmds、gas7、fndc3b、txnrd2、atxn2、cav1/cav2、p16ink4a、six6、abca1、afap1和cdkn2b-as。在治疗青光眼的一些实施方案中,靶序列选自seqidno:143-163中所示的核苷酸序列、或其组合。在一些实施方案中,对主体的施用通过植入、注射(例如,视网膜下)或病毒而发生。
crispr系统可以用于促进真核细胞中的靶向基因组编辑,所述真核细胞包括哺乳动物细胞,例如人细胞。为了促进基因组编辑,将待修饰的细胞与编码cas9或cas9蛋白、dna或rna本身的表达载体,连同指导rna分子本身或包含编码指导rna分子的核酸分子的表达载体共转染。例如,在某些实施方案中,cas9的引入可以通过转染cas9作为蛋白质、rna、dna或包含编码cas9的核酸的表达载体来完成。在某些实施方案中,指导dna本身可以作为rna分子(grna)、dna分子直接施用,或作为包含编码grna的核酸的表达载体施用。
“视网膜变性”意指与神经元或其他神经细胞(例如视网膜神经节或感光细胞)的变性或功能障碍相关的疾病、病症或病况(包括视神经病变)。视网膜变性可以是任何疾病、病症或病况,其中可以发生神经元的功能降低或功能障碍,或者神经元或其他神经细胞的丧失。
此类疾病、病症或病况包括但不限于青光眼、肌萎缩侧索硬化(als)、三叉神经痛、舌咽神经痛、贝尔麻痹、重症肌无力、肌营养不良、进行性肌萎缩、原发性侧索硬化(pls)、假性延髓麻痹、进行性延髓麻痹、脊髓性肌萎缩、遗传性肌萎缩、椎间盘综合症(invertebratedisksyndromes)、颈椎病、丛障碍、胸廓出口破坏综合征、外周神经病变、紫质症、阿尔茨海默氏病、亨廷顿氏病、帕金森氏病、帕金森氏附加性疾病、多系统萎缩、进行性核上麻痹、皮质基底节变性、路易体痴呆、额颞叶痴呆、脱髓鞘疾病、格-巴二氏综合征、多发性硬化、夏-马-图三氏病、朊病毒病、克雅二氏病、gerstmann-straussler-scheinker综合征(gss)、致命性家族性失眠症(ffi)、牛海绵状脑病(bse)、皮克氏病、癫痫和aids痴呆综合症。
其他疾病、病症或病况包括但不限于亚历山大氏病、阿尔珀斯病、共济失调性毛细血管扩张症、贝敦氏症(也称为spielmeyer-vogt-sjogren-batten病)、卡纳万病、科卡因综合征、糖尿病性神经病变、额颞叶变性、hiv相关性痴呆、肯尼迪氏病、克腊伯氏病、神经疏螺旋体病(neuroborreliosis)、马-约二氏病(脊髓小脑性共济失调3型)、湿性或干性黄斑变性、尼曼-匹克二氏病、佩-梅二氏病、光感受器变性疾病如视网膜色素变性和相关疾病、雷弗素姆氏病、桑德霍夫氏病(sandhoff'sdisease)、谢耳德氏病、继发于恶性贫血的脊髓亚急性联合变性、spielmeyer-vogt-sjogren-batten病(也称为贝敦氏症)、脊髓小脑性共济失调(具有不同特征的多种类型)、steele-richardson-olszewski病、脊髓痨、格子状角膜营养不良、视网膜色素变性、年龄相关性黄斑变性(amd)、与湿性或干性amd相关的光感受器变性、其他视网膜变性如视网膜色素变性(rp)、视神经玻璃膜疣、视神经病变和视神经炎如起因于多发性硬化的视神经炎。
可以根据本文公开主题预防或治疗的不同类型青光眼的非限制性实例包括原发性青光眼(也称为原发性开角型青光眼、慢性开角型青光眼、慢性单纯性青光眼和单纯性青光眼)、低眼压性青光眼、原发性闭角型青光眼(又称原发性闭角型青光眼、窄角型青光眼、瞳孔阻滞性青光眼和急性充血性青光眼)、急性闭角型青光眼、慢性闭角型青光眼、间歇性闭角型青光眼、慢性开角闭合型青光眼、色素性青光眼、剥脱性青光眼(也称为假性剥脱性青光眼或囊膜性青光眼)、发育性青光眼(如原发性先天性青光眼和婴儿型青光眼)、继发性青光眼(如炎性青光眼(如葡萄膜炎和福斯氏异色性虹膜睫状体炎))、晶状体源性青光眼(如闭角型青光眼伴成熟性白内障、继发于晶状体囊破裂的晶状体过敏性青光眼、由于晶状体毒性网络阻塞和晶状体半脱位的晶状体溶解性青光眼)、继发于眼内出血的青光眼(如前房积血和溶血性青光眼,又称为红细胞性青光眼)、创伤性青光眼(如房角退缩性青光眼、创伤性前房角后退、术后青光眼、无晶状体性瞳孔阻滞和睫状体阻滞性青光眼)、新生血管性青光眼、药源性青光眼(如皮质类固醇诱发的青光眼和α-胰凝乳蛋白酶性青光眼)、中毒性青光眼以及与眼内肿瘤、视网膜脱离、严重的化学性眼部灼伤和虹膜萎缩相关的青光眼。在某些实施方案中,神经变性疾病、病症或病况是与过度血管生成无关的疾病、病症或病况,例如,并非新生血管性青光眼的青光眼。在一些实施方案中,青光眼的突变靶包括但不限于optn、tbk1、tmco1、pmm2、gmds、gas7、fndc3b、txnrd2、atxn2、cav1/cav2、p16ink4a、six6、abca1、afap1和cdkn2b-as。
如本文使用的,术语“病症”一般而言指获益于用针对鉴定的靶或途径之一的化合物治疗的任何病况,包括可以通过有效量的针对鉴定的靶或途径之一的化合物或其药学上可接受的盐治疗的任何疾病、病症或病况。
如本文使用的,术语“治疗”可以包括逆转、减轻、抑制其进展、预防或减少此类术语适用的疾病、病症或病况的可能性,或者此类疾病、病症或病况的一种或多种症状或表现(例如,引起视网膜神经节或感光细胞功能障碍和/或死亡的疾病或病症)。在一些实施方案中,治疗减少视网膜神经节或感光细胞的功能障碍和/或死亡。例如,与在经历治疗之前的主体或未经历治疗的主体中的视网膜神经节或感光细胞的功能障碍和/或死亡相比,治疗可以使视网膜神经节或感光细胞的功能障碍和/或死亡减少至少5%、10%、15%、20%、25%、30%、33%、35%、40%、45%、50%、55%、60%、66%、70%、75%、80%、85%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%或更多。在一些实施方案中,治疗完全抑制主体中的视网膜光感受器或神经节细胞的功能障碍和/或死亡。如本文使用的,“视网膜神经节或感光细胞”是在视网膜中发现的能够光转导的专门类型的神经元。在一些实施方案中,至少一种基因产物是视紫红质。
在一些实施方案中,在施用于主体之前,将系统包装到单个腺伴随病毒(aav)颗粒内。在一些实施方案中,对主体的施用通过视网膜下注射而发生。治疗、施用或疗法可以是连续的或间歇的。连续治疗、施用或疗法指至少在每天的基础上的治疗,而不中断治疗**或多天。间歇性治疗或施用,或以间歇方式的治疗或施用,指在性质中并非连续的,而是循环的治疗。根据本文公开方法的治疗可以导致疾病、病症或病况的完全缓解或治愈,或者疾病、病症或病况的一种或多种症状的部分改善,并且可以是瞬时的或**的。术语“治疗”还预期涵盖预防、治疗和治愈。
术语“有效量”或“治疗有效量”指足以实现有益或所需结果的药剂的量。治疗有效量可以根据以下一种或多种而变:待治疗的主体和疾病状况、主体的重量和年龄、疾病状况的严重性、施用方式等等,其可以通过本领域普通技术人员容易地确定。该术语还应用于将提供通过本文所述的任何一种成像方法的检测的图像的剂量。具体剂量可以根据以下一种或多种而变:所选择的具体试剂、待遵循的给药方案、它是否与其他化合物组合施用、施用时机、待成像的组织和携带剂量的物理递送系统。
术语“抑制(inhibit)”或“抑制(inhibits)”意指与未治疗的对照主体、细胞、生物途径或生物活性相比,或者与在主体治疗之前主体中的靶相比,降低、压制、减弱、减小、阻止或稳定疾病、病症或病况的发展或进展、生物途径的活性或生物活性例如至少10%、20%、30%、40%、50%、60%、70%、80%、90%、95%、98%、99%或甚至。术语“降低”意指抑制、压制、减弱、减小、阻止或稳定视网膜疾病、病症或病况的症状。应理解,尽管不排除,但治疗疾病、病症或病况并不要求完全消除疾病、病症、病况或与其相关的症状。
如本文使用的,短语“药学上可接受的载体”指药学上可接受的材料、组合物或媒介物,例如液体或固体填料、稀释剂、赋形剂或溶剂包封材料,其涉及从身体的一个器官或一部分携带或转运主题化合物到身体的另一个器官或部分。在与制剂的其他成分相容并且对患者无害的意义上,每种载体必须是“可接受的”。可以充当药学上可接受的载体的材料的一些实例包括:(1)糖,例如乳糖、葡萄糖和蔗糖;(2)淀粉,例如玉米淀粉和马铃薯淀粉;(3)纤维素及其衍生物,例如羧甲基纤维素钠、乙基纤维素和乙酸纤维素;(4)粉末黄蓍胶;(5)麦芽;(6)明胶;(7)滑石;(8)赋形剂,例如可可脂和栓剂蜡;(9)油,例如花生油、棉籽油、红花油、芝麻油、橄榄油、玉米油和大豆油;(10)二醇,例如丙二醇;(11)多元醇,例如甘油、山梨醇、甘露醇和聚乙二醇;(12)酯,例如油酸乙酯和月桂酸乙酯;(13)琼脂;(14)缓冲剂,例如氢氧化镁和氢氧化铝;(15)海藻酸;(16)无热原水;(17)等渗盐水;(18)林格氏溶液;(19)乙醇;(20)ph缓冲溶液;(21)聚酯、聚碳酸酯和/或聚酸酐;以及(22)药物制剂中采用的其他无毒相容性物质。
“药学上可接受的盐”指化合物的相对无毒的无机和有机酸加成盐。
术语“预防(prevent)”、“预防(preventing)”、“预防(prevention)”、“预防性治疗”等等指减少主体中发展疾病、病症或病况的可能性,所述主体未患有疾病、病症或病况,但处于发展疾病、病症或病况的风险中或者易于发展疾病、病症或病况。
术语“主体”和“患者”在本文中可互换使用。在其许多实施方案中通过本文公开方法治疗的主体期望地是人主体,尽管应理解本文所述的方法关于所有脊椎动物物种都是有效的,所述脊椎动物物种预期包括在术语“主体”中。相应地,“主体”可以包括用于医学目的人主体,例如用于治疗现有病况或疾病或者用于预防病况或疾病发作的预防性治疗,或者用于医学、兽医学目的或发育目的的动物主体。合适的动物主体包括哺乳动物,包括但不限于灵长类动物,例如人、猴、猿等等;牛科动物,例如牛、黄牛等等等;绵羊动物,例如绵羊等等;山羊动物,例如山羊等等;猪科动物,例如猪、阉公猪等等;马科动物,例如马、驴、斑马等等;猫科动物,包括野猫和家猫;犬科动物,包括犬;兔类动物,包括兔、野兔等等;以及啮齿类动物,包括小鼠、大鼠等等。动物可以是转基因动物。在一些实施方案中,主体是人,包括但不限于胎儿、新生儿、婴儿、青少年和成人主体。此外,“主体”可以包括患有或怀疑患有病况或疾病的患者。
术语“疗效”指由药理学活性物质引起的在动物,特别是哺乳动物,且更特别是人中的局部或全身效应。
如本文使用的,术语“治疗有效量”和“有效量”意指本发明的组合物的量,其以适用于任何医学治疗的合理的利益/风险比,在动物中的至少细胞亚群中有效产生一些所需疗效。
iii.一般定义
尽管本文采用了特定术语,但它们仅以一般性和描述性意义使用,而不是出于限制性目的。除非另外定义,否则本文使用的所有技术和科学术语都具有与本文所述主题所属领域的普通技术人员通常理解相同的含义。
遵循长期存在的**法惯例,在本申请包括权利要求中使用时,术语“一个”、“一种”和“该/所述”指“一个或多个/一种或多种”。因此,例如,对“主体”的提及包括多个主体,除非上下文明显相反(例如,多个主体),等等。
在说明书和权利要求自始至终,术语“包含(comprise)”、“包含(comprises)”和“包含(comprising)”以非排他性的意义使用,除非上下文另有要求。同样地,术语“包括”及其语法变体预期是非限制性的,使得列表中的项目的叙述不排除可以取代或加入所列项目的其他类似项目。
为了本说明书和所附权利要求的目的,除非另有说明,否则表示说明书和权利要求中使用的量、大小、尺寸、比例、形状、制剂、参数、百分比、参数、数量、特征和其他数值的所有数目,应理解为在所有情况下都用术语“约”进行修饰,即使术语“约”可能没有明确地出现在值、量或范围处。相应地,除非有相反的指示,否则在下述说明书和所附权利要求中阐述的数值参数不是**的并且无需是**的,而是可以根据需要近似和/或更大或更小,其反映公差、转换因子、四舍五入、测量误差等等以及本领域技术人员已知的其他因素,这取决于寻求通过本文公开主题获得的所需性质。例如,当提及值时,术语“约”可以意欲涵盖与指定量在一些实施方案中±、在一些实施方案中±50%、在一些实施方案中±20%、在一些实施方案中±10%、在一些实施方案中±5%、在一些实施方案中±1%、在一些实施方案中±0.5%、以及在一些实施方案中±0.1%的变化,因为此类变化适于执行所公开的方法或采用所公开的组合物。
此外,当与一个或多个数目或数值范围结合使用时,术语“约”应理解为指所有此类数目,包括范围中的所有数目,并且通过扩展所述数值上方和下方的边界来修改该范围。通过端点的数值范围的叙述包括所有数目,例如包含在该范围内的整数,包括其分数(例如,1至5的叙述包括1、2、3、4和5,及其分数例如,1.5、2.25、3.75、4.1等等),以及该范围内的任何范围。
例证
已包括下述实施例,以对本领域普通技术人员提供了用于实践本文公开主题的代表性实施方案的指导。鉴于本公开内容和本领域的一般技术水平,技术人员可以了解,下述实施例仅预期是示例性的,并且可以采用众多变化、修改和改变,而不脱离本文公开主题的范围。下述合成描述和具体实施例仅用于说明性目的,并且不应解释为以任何方式限制通过其他方法制备本公开内容的化合物。
方法
将人胚胎肾(hek)细胞系293t(lifetechnologies,grandisland,ny)在37℃与5%co2/20%o2下维持在达尔贝科改良伊格尔培养基(dmem)(invitrogen)中,所述dmem补充有10%胎牛血清(gibco,lifetechnologies,grandisland,ny)和2mmglutamax(invitrogen)。
靶向视紫红质的grna(参见ranganathan,v和zack,dj.grid:acrisprguidernadatabaseandresourceforgene-editing.submitted(2015))通过重叠寡核苷酸生成,所述寡核苷酸通过pcr使用两步扩增phusionflashdna聚合酶(thermofisherscientific,rockford,il)进行退火且扩增,并且随后使用zymodna清洁和浓缩柱进行纯化。然后将纯化的pcr产物重悬浮于h2o中,并且使用nanodrop1000(thermofisherscientific)定量。使用gibsonassembly(newenglandbiolabs,ipswich,ma)(gibson等人naturemethods6:343-345(2009))伴随略微修改生成表达grna的构建体。总反应体积从20μl减少到2μl。通过桑格测序验证克隆。
用cas9(未修饰的或细胞周期调节形式)和靶向视紫红质的grna构建体共转染hek293细胞。转染后48小时,收获基因组dna,并且根据附录中列出的引物扩增靶切割位点周围的序列。然后在执行t7endoi测定之前纯化且定量pcr产物。简言之,使200ngpcr产物变性,然后缓慢再退火以允许形成异源双链体,将t7核酸内切酶i加入pcr产物中,并且在37℃下温育25分钟以切割异源双链体,反应在上样染料中淬灭,并且更后,在6%tbepage凝胶上运行反应以分辨产物。凝胶用sybr-gold染色,显现,并且使用imagej定量。使用二项式导出的方程计算nhej频率:
;其中“a”和“b”的值等于在本底扣除后的切割片段的积分面积,并且“c”等于在本底扣除后未切割的pcr产物的积分面积。
实施例1
虽然仍处于起步阶段,但crispr系统已彻底改革了基因组编辑技术,改变了生物学研究,并且开创了遗传医学的新时代。针对人基因组中的序列,可以定制基于crispr的编辑系统,以破坏任何基因或调节元件,或以高度特异性形式缺失且替换基因组dna序列。并且尽管跨越全世界的众多研究已证实疾病突变可以在体外有效靶向,但用于体内使用的基于crispr的治疗剂的开发已被递送约束显著阻碍。
腺伴随病毒(aav)载体是基因治疗中更频繁使用的病毒载体,具有几个吸引人的特点:病毒是非致病性的,它感染分裂细胞和非分裂细胞两者,表达可以持续很长一段时间,以及在临床试验中安全性、功效和毒性的一般缺乏的值得注意的历史。另外,变体aav血清型的组合在靶向特定细胞类型方面是有效的。虽然aav载体提供了递送治疗性crispr组件的安全手段,但存在限制其效用的一个主要技术障碍–其大小。野生型aav基因组为长度~4.7kb,并且重组病毒可以包装至多5.0kb。该包装容量限定了可以用于单个病毒载体的dna的上限。
crispr/cas9系统由核酸酶(cas9)和指导rna(grna)构成,其作用于将核酸酶导向基因组中的特定区域。更常用的cas9蛋白来自化脓性链球菌(spcas9),其由4104bp的开放读码框编码。已假设由于spcas9的大尺寸,两种crispr组件,包括表达所必需的启动子和终止子序列的递送,受aav包装容量限制。恰当地使用标准启动子元件,spcas9和grna盒以显著余量超出aav的包装容量。
在wo2015/195621(通过引用整体并入本文)中公开了包装容量问题的解决方案,其极大地扩展了关于治疗性crispr的应用的潜在范围。crispr递送的这种新方法的核心是在人细胞中高度活性的紧凑双向启动子。h1启动子是非常独特的遗传元件,其被rna聚合酶polii和poliii识别。引入aav载体内,h1启动子可以有效地表达cas9和grna两者。因为优化的h1启动子长度仅为~230bp,所以该盒的使用允许在单一重组aav中包装spcas9和多种杂合grna。
包含spcas9、短多聚a(spa)序列、两个可分开定制的grna支架和h1启动子元件的完整的“一体化”aav载体是~4640kb。这几乎是野生型aav基因组的大小,并且仍然远低于5.2kb的更大包装容量。任何cas9基因都可以掺入这种基本结构内。该平台因此提供了比任何现有技术在体内靶向多得多的基因组位点和突变的能力(图1)。
由来自金黄色葡萄球菌(s.aureus)的cas9(sacas9)突出显示的用于临床递送的另一种可行方法是使用可以包装到aav载体内的较小的cas9直向同源物。虽然替代的cas9蛋白具有替代的靶向特异性,并且可能比spcas9蛋白更具限制性,但与h1系统组合的较小的sacas9蛋白的使用提供了超过现有方法的显著优点。
crispr-cas9系统通过诱导dsdna断裂起作用,然而,cas9中的单点突变可以生成ssdna“切口酶”。虽然切口酶的使用需要两倍的grna来生成dsdna断裂,但它被普遍公认为更安全。重要的是,aav-h1-crispr平台可以容纳sacas9和超过4种grna,并且因此可以安全地生成dna断裂,而无脱靶突变。目前的递送方法缺乏这种能力。
利伯氏先天性黑蒙(lca)由一组早发型儿童视网膜变性构成,其特征在于在生命的前几个月期间严重的视网膜功能障碍和严重的视力损害或失明。lca,作为一种罕见疾病,是遗传性失明的更常见原因,构成所有已知遗传性视网膜变性疾病的多达5%。具体地,lca10–对于其不存在fda批准的治疗的一种疾病–由cep290基因中的内含子突变引起。这种a至g突变导致产生从头的强剪接供体位点,以及隐藏外显子(外显子x)包含在cep290mrna内和后续光感受器或神经节变性。该突变作为治疗靶特别有吸引力。
成功证实原型aav-h1-crispr在人细胞中稳固地表达cas9和grna两者,并且从而可以在体外有效地指导基因靶向。使用视网膜疾病的小鼠模型,证实h1-aav-crispr可以用于通过视网膜下递送**地靶向体内致病性突变(图2)。
用crispr治疗lca10的潜在原因的基于aav的策略目前处于开发中,用于通过editasmedicine的临床使用。他们解决aav包装容量问题的方法是采用来自金黄色葡萄球菌的较小的cas9直向同源物(sacas9),其由~3.2kb转录物编码。sacas9基因的紧凑大小允许将其连同两个grna盒一起包装到单个aav载体内,然而我们的aav技术提供了可以用于另外grna的另外空间。
就基于crispr的治疗剂的安全性问题而言,更重要的无疑是脱靶诱变;如果cas9在非预期的位置处切割dna,则这可能发生。幸运的是,通过采用仅切割一条dna链的修饰形式的cas9(称为d10a切口酶),几乎可以消除这种风险。通过分别接合两个grna以在相反链上生成两个紧密相对的切口,cas9切口酶可以有效地生成双链断裂。然而,脱靶断裂只有在两个grna识别在基因组中其他地方紧密接近且在相反链上存在的脱靶时通过切口酶生成,这是统计上不大可能的(churchg.nature2015dec3;528(7580):s7)并且因而未观察到的事件;带切口的dna被有效修复而细胞蛋白质是正常的。出于这个原因,引入具有四个grna(每侧上一对以缺失靶向突变)的cas9(d10a)用于校正cep290突变被普遍和明确地视为更安全的方法-包括在监管机构中。(shen等人naturemethods11,399–402(2014))。h1元件独特地赋予使用该容量的空间。
然而,由于目前系统的尺寸限制(即editas:具有非h1系统的sacas9),cep290突变的校正(其涉及丢弃大约1kb的内含子dna)不能通过更安全的切口酶方法来完成。已鉴定了允许我们将更安全的治疗剂递送至临床的四种grna:
然而,由于u6启动子,editas已将其靶位点约束至5'g靶向位点。(friedland等人genomebiology16:257(2015))。
存在与a至g突变重叠的spcas9靶向位点。使用先前公开的h1系统和spcas9,可以直接靶向cep290突变而无需生成大缺失。该方法还预期比目前的editas系统更安全。
来自5'g起始要求的减少位点示出了相对于u6在使用h1表达grna中的效用。另外,靶向位点的总数目显示spcas9在数目上超过sacas9,如计算预测的(ranganathan等人natcommun2014aug8;5:4516)。
实施例2
本文提供的是基于aav的方法以治疗lca。与涉及基因转移的治疗形成对比,该方法使用crispr基因组编辑技术。crispr近年已得到了发展,并且已非常迅速地彻底改革了生物学研究,并且开创了遗传医学的新时代(4,5)。crispr由细菌核酸内切酶和短rna构成,所述短rna将该核酸酶引导至基因组中的特定切割位点。利用定制的指导rna(grna),crispr可以进行编程,以破坏任何人基因或调节元件,或以高度特异性形式缺失且替换基因组dna序列。针对lca的crispr方法将促进从处于变性风险中的细胞中**去除突变,并且因此治愈该疾病。
采用aav用于crispr应用存在一个主要的技术障碍–其大小。aav是可以包装至多5.2kbdna的小病毒。标准crispr以显著余量超出这种包装容量。crispr由细菌核酸内切酶cas9和至少一种grna构成。来自化脓性链球菌的更常用的cas9蛋白由4.1kb基因单独编码。将两种crispr组件–表达所必需的标准启动子和终止子序列–包装到单一病毒内是不可能的。
通过引用并入本文的wo2015195621公开了包装容量问题的解决方案。crispr递送的这种新方法的核心是称为h1的紧凑双向启动子。单个h1启动子可以有效地表达cas9和grna两者。这种独特的遗传元件允许由任何cas9基因和多个grna(更佳为四个)构成的组装包装在单个重组aav(图1)中,称为aav-h1-crispr。关于添加grna的容量允许aav-h1-crispr生成具有不匹配的位点特异性的双链断裂,因此使众所周知的脱靶诱变风险更小化(6,7)。
本文提供的是通过单独施用的aav-h1-crispr治疗剂用于lca10的安全且持久的治疗。lca10由基因cep290中的突变引起。这种lca亚型影响了西方世界的约2,000位个体。这种治疗剂与由editasmedicine正在开发的目前技术形成对比。他们对包装容量问题的解决方案是采用较小的cas9基因。然而,它们的载体构型可以接合比我们更少的基因组靶,并且缺乏关键特点:使用四种grna以提供精巧的靶向灵敏度(如上文提及的),其已显示防止脱靶诱变,这是重要的安全问题(6,7)。aav-h1-crispr保留了这个关键特点。通过我们的lca10载体引起的脱靶突变率预期是可忽略不计的。
1.病毒构建体的组装。为了促进在商业实验室背景下来自模块组件的病毒载体的快速组装,将合成合成模块盒,其将可重复用于后续靶。用于lca10的载体将由这些一般模块和四个cep290特异性h1-grna模块组装。更终的组装将通过限制性酶切作图然后通过完整测序加以验证。更终产物将作为1mg转染级cep290特异性aav穿梭质粒dna的组装病毒构建体(不含内毒素和序列错误)提供。
2.aav-h1-crispr原种的制备和分析。病毒构建体的包装、病毒颗粒的纯化和病毒滴度纯度和感染性的分子评价由cgmp认证的核心设施根据行业标准(10)执行。生产定性和定量上适合于临床前测试的病毒原种。cgmp制备和纯化的无菌、无内毒素aav的原种,具有0.9iu/病毒基因组的更小感染率、1012个病毒基因组/ml的更小滴度和1014个感染单位的更小产率。
3.aav-h1-crispr基因组靶和脱靶位点的定量表征。使用永生化的视网膜色素上皮细胞系htert-rpe1进行功能验证,其具有lca10靶位点。受感染的细胞群体中的中靶(on-target)和预测的脱靶位点将进行深度测序。修饰/未修饰的等位基因比率将提供效率的定量量度;中靶/脱靶修饰比率将是特异性的确定性量度。使用改造的小鼠和人活组织检查样品在体内测试病毒。靶接合的定量分析。可以在培养物中产生>5%的cep290等位基因中的缺失的病毒(5%的阈值被认为导致体内视力改善),具有<0.1%的脱靶效应。
1.maguiream等人nengljmed.2008;358(21):2240-2248.
2.schimmerj等人humgenetherclindev.2015;26(4):208-210.
3.azvolinskya.natbiotechnol.2015;33(7):678-678.
4.barrangour.science.2014;344(6185):707-708.
5.barrangour等人expertopinbiolther.2015;15(3):311-314.
6.shenb等人natmethods.2014;11(4):399-402.
7.churchg.nature.2015;528(7580):s7.
8.philippidisa.humgenetherclindev.2015;26(2):1-4.概述于近期的新闻报道:http://www.reuters.com/article/us-sparktherapeutics-study-idukkcn0rz0wx20151005
9.touchotn,flumem.natbiotechnol.2015;33(9):902-904.
10.wrightjf.genether.2008;15(11):840-848。
实施例3
本文提供的是通过开发crispr/cas9基因编辑技术来治疗adrp的替代方法(doudna,j.a.等人science346,1258096(2014);hsu,pd.,等人cell157,1262-1278(2014)),其中rna引导的核酸内切酶与可定制的小指导rna(grna)结合使用,以靶向且切割突变型视紫红质等位基因,其通过易错的非同源末端连接(nhej)特异性地敲除表达突变型等位基因,而不影响正常等位基因。尽管这种方法可能导致野生型水平的视紫红质的仅50%的表达,但动物数据提示这应该足以提供临床上有用的视杆功能(liang,y.等人thejournalofbiologicalchemistry279:48189-48196(2004))。
视紫红质中的r135突变导致更具侵袭性和快速进展形式的视网膜色素变性(rp)。受影响的个体,在儿童期过程中具有夜盲症,且在生命的第二个十年之前视野丧失。疾病进展异常高,具有每年基线erg幅度和视野面积的平均50%丧失。到生命的第四个十年,黄斑功能严重受损(omim:http://www.omim.org/entry/180380)。
通过一些估计,r135突变占全球第二常见的视紫红质突变。r135突变特别顺应通过nhej的校正,因为过早终止密码子可能通过无义介导的衰变导致降解的转录物,减轻该突变的显性负效应。更普遍的突变p347在视紫红质的外显子5中发生,这对于通过crispr基因组编辑的校正提出了另外的挑战。基因的更后一个外显子中的过早终止密码子不易受无义介导的衰变的影响。
1.audoi等人investophthalmolvissci.(2010)jul;51(7):3687-700.
2.pannaralemr等人ophthalmology.(1996)sep;103(9):1443-52.
3.andréassons等人ophthalmicpaediatrgenet.(1992)sep;13(3):145-53。
crispr靶位点
wt序列靶向:
r135g靶向:
r135w靶向:
r135l靶向:
grna序列:
实施例4
青光眼,全世界不可逆性失明的首因(1),是视神经病变,其中在视神经头的筛板处的视网膜神经节细胞(rgc)轴突的进行性损伤导致轴突变性和细胞死亡(2)。目前,**的治疗,无论是通过滴眼液、激光还是切口手术,都是降低眼内压(iop)并且减少在视神经头处的损伤。不幸的是,这在一些患者中是困难的,而在其他患者中,尽管侵袭性iop降低,该疾病仍可继续恶化。该领域长期以来需要替代治疗策略,所述治疗策略能够通过减轻对残余轴突损伤的rgc应答来补充iop降低。此外,nei已将视神经再生列为其大胆的目标,并且任何再生疗法都必须解决视神经切断的rgc存活的问题。为此,非常需要开发可能直接干扰rgc轴突变性和/或轴突损伤相关细胞死亡的活跃遗传程序的神经保护剂(3-6)。
为了以全面和无偏方式鉴定可以充当关于神经保护性青光眼治疗的新药物靶的基因,在整个小鼠激酶组(基因组的可药物靶向子集(druggablesubset))中筛选其抑制促进rgc存活的激酶。对于该筛选,开发了高通量方法用于敲低来自雄性和雌性c57bl/6小鼠的原代rgc中的基因表达(7),其使用小干扰rna寡核苷酸(sirna)并且将其与rgc存活的定量测定偶联(celltiter-glo,promega)(5)。使用这种方法,就免疫淘选后三天(其必须将细胞视神经切断)增加rgc存活的能力,筛选了靶向623种激酶的1869个sirna的阵列文库。前两个命中是双亮氨酸拉链激酶(dlk/map3k12)和其**已知的底物,丝裂原激活蛋白激酶激酶7(mkk7)。使用雄性和雌性floxeddlk小鼠(8)和表达cre、衣壳修饰的(9,10)、腺伴随病毒(aav2)的玻璃体内注射,显示dlk的靶向破坏致使rgc在小鼠视神经挤压模型中对轴突损伤诱导的细胞死亡高度抗性(5)。此外,因为junn末端激酶(jnk)1-3(通常由一种或多种上游map3k激活)已显示在rgc细胞死亡中发挥关键作用(11,12),所以测试了dlk(map3k12)是否是相关的上游触发器。实际上,然而用aav2-cre玻璃体内注射的野生型小鼠响应视神经损伤且具有rgc体细胞和轴突中jnk信号传导的强烈上调,floxeddlk小鼠具有该途径的减少激活(5)。更后,使用公布的激酶抑制剂概况分析数据(13,14),蛋白激酶抑制剂陶扎色替被鉴定为dlk的抑制剂,并且显示它在大鼠视神经横切和青光眼模型两者中保护rgc(5)。在一些实施方案中,关于dlk/map3k12的靶序列可以包含选自seqidno:788-1023的核苷酸序列。
注意到陶扎色替在体外一致地改善rgc存活多于单独的dlk敲低或敲除(5)。这提示陶扎色替的一种或多种另外的激酶靶(其中存在多于150种)可能与dlk合作以促进细胞死亡,并且两者的同时抑制促进更大rgc存活。为了鉴定这些其他靶,修饰激酶组筛选以在每个孔中包括dlksirna,并且使其对与dlk敲低协同合作的那些文库sirna致敏,以进一步增加rgc存活(图19a)。另一种修饰是在用秋水仙碱加重免疫淘选损伤之前,允许48小时的现存蛋白质周转,所述秋水仙碱是已用于体外和体内建模轴突损伤的微管去稳定剂(15,16)。如预期的,因为dlksirna已经存在于任何地方,所以来自文库的dlksirna寡核苷酸(其在初始激酶组筛选中更有效)无一改善存活高于基线。相反,该修饰的“协同作用”方案中的**基因是紧密相关的混合谱系激酶,亮氨酸拉链激酶(lzk/map3k13)。具有在初始筛选中未使用的序列的另外四种lzksirna确认lzk敲低(图19b,插图)本身具有很少的活性,但在促进原代rgc存活方面与dlk敲低高度协同合作(图1b)。此外,这种协同作用对lzk是特异性的,因为混合谱系激酶家族(即mlk1-3)的其他成员无一对存活具有任何作用(数据未显示)。因为除dlk之外,陶扎色替的确还抑制lzk(13,14),所以考虑lzk是否实际上是“其他”关键靶。与该假设一致,lzk或dlk的各个敲低是部分神经保护性的,并且使细胞对低剂量的陶扎色替致敏(如将预期的,如果关键药物靶已经被遗传破坏)。此外,dlk和lzk两者的组合敲低重现了通过药物产生的存活,并且陶扎色替的进一步添加没有作用(如将预期的,如果所有关键药物靶已经被遗传破坏)(图19c)。在一些实施方案中,关于dlk/map3k13的靶序列可以包含选自seqidno:1024-1397的核苷酸序列。
虽然关于lzk作为rgc细胞死亡的关键介质的证据是实质性的,但它完全基于各个sirna。值得注意的是,通过任何给定sirna产生的表型是由所有21个核苷酸介导的经典“中靶”沉默以及由六至七核苷酸种子序列介导的混杂的“脱靶”沉默的生物学总和(17,18)。后者可以使数百个靶沉默,不幸的是它导致显性表型和sirna筛选结果(19)。因此,选择两种互补的方法以验证lzk发现,sipool和成簇规律间隔短回文重复(crispr)介导的基因敲除。sipool是靶向共同基因的30种sirna的库。因为30种组件sirna中的每一种都具有不同的脱靶组,但共享共同的中靶,所以sipool对于中靶相对于脱靶沉默提供了30倍的富集(20)。如预期的,虽然lzksipool仅更低限度地改善了原代rgc存活,但dlk和lzksipool的组合在存活(图19d)和jnk信号传导的压制(图19e)两者方面是高度协同的。表型不是celltiter-glo测定的人为现象,因为使用常规活力染剂获得类似结果的(图19f)。对于使用crispr的敲除方法,利用了主要rgc平台围绕小rna(即sirna)的递送构建的事实。通过从在rosa26基因座处表达化脓性链球菌cas9(spcas9)的小鼠分离rgc(21),并且相反用体外转录的合成指导rna(sgrna)转染,可以靶向cas9以在给定的靶基因中切割。然后细胞用非同源末端连接修复该切割位点,产生小的(~1-20bp)插入/缺失(插入缺失),其可以移码下游蛋白质。为了验证该方法,合成了针对dlk的一系列sgrna,并且显示它们与lzksipool协同合作以增加存活(图19g),并且它们产生靶特异性插入缺失而没有可检测的脱靶效应(图19h)。交互方法,即dlksipool和lzksgrna,产生了与仅使用sgrna,即dlksgrna和lzksgrna(图19j)相似的结果(图19i)。更近生成了floxedlzk小鼠并且验证了体外敲除(图19k)。对纯合的dlkf1/fllzkfl/fl小鼠进行育种且进行测试,以察看lzk和dlk的体内组合靶向破坏是否导致视神经损伤后增强的rgc存活和jnk途径压制。然而,至少在原代rgc中,清楚的是,当dlk无功能时,lzk可以介导损伤信号传导,需要dlk和lzk两者的同时抑制用于更大的神经保护。
接下来寻求鉴定损伤信号传导途径的非激酶成员,并且因此我们扩展了我们的平台以筛选16,877个sirna微型库的全基因组文库(每种基因四个)。为了改善测定的信噪比并且促进在全基因组规模下的筛选,利用lzk抑制对改善基线存活几乎没有作用,但使细胞对进一步dlk抑制高度致敏的事实。因此,除标准文库sirna微型库之外,每个孔也接受lzksipool。为了分析结果,利用基因组规模筛选多次取样具有每种可能的种子组合的sirna的事实,因此允许两种不同的生物信息学方法来解决普遍的脱靶效应。首先,可以跟踪给定种子的存活效应,因为它在文库自始至终出现了数十次。然后,这可以用于生成校正因子,其帮助扣除对表型的脱靶贡献并且并且揭示由中靶介导的组件(19)。其次,除已知通过每个种子产生的存活/毒性之外,还可以预测所有其可能的脱靶。然后,使用haystack分析通过类似行为的种子搜索共同靶向的基因,事实上可以尝试解卷积(deconvolute)脱靶效应并且鉴定其脱靶沉默影响存活的基因(22)。
当分析使用**种方法(种子校正的,中靶)的结果时,发现dlk是所有16,877种基因中的**基因(图20a)。此外,mkk4、mkk7、jnk1、jnk2、jnk3都排序在前1%中(数据未显示)。该筛选还鉴定了两种已知的jnk依赖性转录因子,jun和atf2。前者先前已被验证为视神经损伤的啮齿类动物模型中rgc细胞死亡的关键介质(23)。为了测试后者的作用,从野生型相对于floxedatf2小鼠中分离原代rgc(24),并且用cre表达相对于gfp表达的腺病毒转导它们。如预期的,atf2的靶向缺失增加存活,其与通过dlk缺失产生的那种相当(图20b)。总之,这些结果提供了筛选能够鉴定已知dlk/jnk途径成员的信心。接下来,尝试验证新型基因,其与dlk或jnk没有先前关联。使用haystack分析,**命中是性别决定区y(sry)-盒11(sox11)转录因子。连同sox4一起,sox11先前已显示对于在发育过程中的rgc存活和分化是关键的(25)。为了验证sox11在rgc细胞死亡中发挥矛盾作用,首先测试sox11sipool并且发现它实际上与lzk(图20c)或dlk(数据未显示)sipool合作,以增加原代rgc存活。检查两种敲除模型:用cre表达的腺病毒相对于gfp表达的腺病毒转导sox11fl/flrgc(26)(图20d),并且用spcas9-敲入rgc与靶向单个sox11外显子的sgrna转染(图20e)。在这两种情况下,sox11的靶向破坏改善了rgc存活。这种发育存活基因可以在成年人的损伤信号传导中发挥作用这一事实通过微阵列研究得到支持,所述微阵列研究已显示sox11在响应视神经挤压的更高上调的基因中,并且以几乎完全是dlk-依赖性的方式(6)。sox11fl/fl小鼠目前用于验证体内存活表型。为了确保潜在的rgc细胞死亡途径成员没有丢失(因为筛选过度敏感以响应dlk途径抑制),在不存在lzksipool的情况下筛选不同的全基因组sirna文库。用靶向17,575种基因的52,725个sirna的阵列文库转染原代rgc。再次,**命中包括已知基因,如dlk、jun、atf2、mkk4和mkk7。然而,这次我们还鉴定了另一种转录因子,肌细胞增强因子2a(mef2a),其在神经元存活和分化中具有突出作用(27-29)。与其他候选物一样,mef2a用sipool(数据未显示)、sgrna(数据未显示)和通过来自条件敲除(mef2afl/f)且用adeno-cre相对于-gfp转导的小鼠的分离rgc(30)进行验证(图20f)。此外,体内mef2a敲除证实在小鼠视神经挤压模型中保护rgc(图20g)。更后,使用dlkfl/fl小鼠,显示mef2a以dlk依赖性方式响应轴突损伤而磷酸化(图20h)。
到此为止,鉴定了四种不同的转录因子(即atf2、jun、sox11、mef2a),每种转录因子在破坏时具有部分保护效应。鉴于来自dlk和lzk的冗余经验,考虑这四种转录因子可能需要被同时抑制用于更大神经保护的可能性。因此,用单独或组合的mef2a、jun、sox11或atf2sipool转染野生型rgc,并且在96小时后测量存活。结果显示四种转录因子的抑制是高度协同的,增加与同时dlk/lzk抑制一样多的存活(图21a)。为了确定这四种转录因子是否在dlk的遗传下游,用dlk/lzksipool连同或不连同靶向四种转录因子的sipool转染野生型rgc。两天后,用腺病毒(表达小鼠sirna不敏感性,大鼠dlk)重构dlk信号传导,并且在另外两天后测量存活。而在四种转录因子的存在下,80-90%的rgc通过dlk过表达杀死,mef2a、sox11、jun和atf2的同时敲低完全消除了细胞死亡(图21b)。更后,使用jnk1-3和mkk4/7敲除和dlk(数据未显示)或lzk过表达(图21c)的组合,证实dlk/lzk在不存在完整mkk/jnk途径的情况下没有毒性效应。总之,功能基因组筛选可以通过模型来概括,其中轴突损伤触发dlk/lzk、mkk4/7和jnk1-3的激酶信号传导级联,导致四种转录因子mef2a、sox11、jun和atf2的激活以及更终的细胞死亡。
靶向dlk作为用于青光眼的神经保护策略具有几个吸引人的特点。如上文提及的,它具有稳固的进化上保守的表型(5,31),它使用无偏方法鉴定,并且很容易用小分子抑制剂药物靶向。此外,它已独立地进行验证,并且dlk抑制而不是简单地延迟细胞死亡,看起来完全预防它(6)。此外,具有dlk抑制保持存活的细胞在体外保持电生理活性(5),并且在体内保留相对健康的基因表达模式,尽管有它们之前的损伤(6)。更后,一些(4,6,8)但并非全部(32)数据提示dlk在由轴突损伤触发的轴突变性程序中起作用。另一方面,dlk抑制延缓轴突再生(6),这是关于神经再生策略的生成的潜在限制(33-35)。这强调了从负责轴突再生的那些中剖析出负责细胞死亡决定的途径成员的需要。此外,尽管它们在细胞死亡(和再生)中的关键作用,但dlk和lzk通过其响应轴突损伤而激活的机制以及它们通过其激活下游转录因子的机制尚未阐明。
如本文公开的,功能基因组筛选与crispr/aav治疗平台整合,以便进一步探测dlk/lzk细胞死亡途径,并且用活基因治疗载体无缝地制止各种候选神经保护靶的活性。
在一些实施方案中,使用sgrna和sirna网络的基于rna的筛选范例的增强集合将用于鉴定新型dlk/lzk途径成员,包括尚未鉴定的上游激活物和靶,其可能解离细胞死亡和再生。在一些实施方案中,开发crispr/aav载体,其允许在视神经病变的啮齿类动物模型中快速验证来自sa1的命中。创建具有适合于基因治疗应用的这些神经保护病毒的变体,从而扩展潜在靶的空间以包括药物不可靶向的基因产物和甚至基因的网络。在一些实施方案中,蛋白质组学和功能丧失/功能获得实验的组合将用于探测dlk通过其调节下游介质mef2a的机制,并且确定mef2a抑制是否可以充当促进rgc存活且不阻止再生的治疗策略。
从机制的观点来看,筛选平台将疾病相关的原发神经元(即rgc)与可以在几周内完成的阵列的、全基因组规模、基于rna的筛选独特地组合,以给予我们无偏且综合的工具,以更好地理解rgc细胞死亡信号传导。实际上,对于rgc发育关键的sox11是此类方法可以如何进行关于涉及细胞死亡的基因的新型和意外发现的完美实例。下面提出的修改(即筛选基因网络和使用crispr技术)将打开可以鉴定的潜在神经保护靶的空间。
从治疗的观点来看,rgc中的crispr编辑将用于开发体内aav/crispr治疗剂。在一些实施方案中,两个分开的顺反子(即,一个表达grna而另一个表达spcas9)的aav/crispr包装到单个aav颗粒内,克服了crispr用于基因治疗的用途的主要局限性。这产生了关于基于基因疗法的方法,以急剧改善的流通量验证来自我们筛选的命中,并且能够在治疗上靶向这些途径成员(包括用小分子不容易药物靶向的那些)的潜力。总体而言,dlk靶途径本身和方法两者构成了用于开发青光眼和其他形式的视神经疾病的新型神经保护疗法的创新策略。
在原代rgc中使用增强的基于rna的筛选来鉴定新型dlk/lzk途径成员,包括尚未鉴定的上游激活物。
dlk和lzk通过其响应视神经损伤而激活的机制仍有待确定。在黑腹果蝇(d.melanogaster)和秀丽隐杆线虫(c.elegans)中,e3连接酶(分别称为highwire和rpm-1)压制dlk同源物(分别称为wallenda和dlk-1)的基础蛋白水平。响应轴突损伤,这种压制是松弛的,导致dlk积累和细胞死亡。有趣的是,关于highwire/rpm-1同源物在小鼠中的作用phr1较不明确。如预期的,背根神经节细胞中phr1的破坏增加了dlk的水平(36,37),但在rgc中,phr1的敲低对dlk水平或存活没有作用(数据未显示),并且有条件的phr1敲除小鼠具有正常rgc存活(38,39)。秀丽隐杆线虫dlk-1中描述的固有钙敏感基序(40)看起来不在功能上作为内源性小鼠lzk(含有该基序的**小鼠dlk-1同源物)的替代物保留,其中野生型人lzk或缺乏六肽基序的突变体导致等价的细胞死亡(图4)。更后,在脊椎动物中,存在几个dlk磷酸化位点,包括s643、s302、s295、s302、t306和s643,其看起来不受已知的dlk激酶(即jnk和dlk)调节,提示新型、上游调节激酶的可能性(37)。虽然上述的激酶组和基因组筛选致敏以寻找dlk途径成员,但dlk/lzk上游的任何基因尚未被鉴定。值得注意的是,这些筛选具有两个固有局限性。首先,敲低而不是敲除,可能不足以产生关于某些基因的表型。支持这一想法,已显示当与基于敲除的筛选直接比较时,基于敲低的筛选可以产生不同的命中(41)。其次,与dlk/lzk、mkk4/7、jnk1/2/3和sox11/atf2/jun/mef2a的情况一样,可能需要同时抑制给定层级的多个成员,以便产生稳健的表型。为了解决这些潜在的局限性,进行对我们的筛选策略的两个关键改变。对于我们的初始重新筛选,鉴于其相对较小的尺寸、生物活性和药物靶向性,将集中于激酶组。
1.筛选靶向小鼠激酶组的阵列sgrna文库,以鉴定其靶向破坏改善rgc存活的基因。
为了解决上文提及的**个局限性,将执行基于crispr的筛选,多少类似于icrispr系统(42)。当我们从这些小鼠中分离rgc并且用sgrna而不是sirna转染它们时,标定了表达spcas9的小鼠的群体并且证实了稳健的存活(图19g-j)。实际上,z'在0.3-0.5之间,其中0.5指示在用阳性对照sgrna(即靶向dlk)转染的筛选中的细胞具有的存活比用对照tracrrna(其缺少将其导向特定基因的20核苷酸的前间区序列)转染的细胞高12个标准差。如图19g和j中所示,靶向相同基因的不同sgrna中的可变性相当小,并且每种基因的三个sgrna应该具有足够的覆盖。与对于dlk和lzk完成的一样,将选择具有序列gn19ngg的靶位点,以便与t7-转录和spcas9、前间区序列邻近基序(pam)相容。将优先考虑生物信息学预测为具有更大的中靶和更低的脱靶切割的那些位点(43-45)。此外,在基因的5'一半中,并且在可能时,在关键结构域中选择靶位点。前者确保通过插入缺失产生的移码突变影响剩余蛋白质的多于一半,而后者允许留在框内的三分之一的插入缺失仍然破坏蛋白质功能。为了允许与过去工作的直接比较,该文库将靶向sigma激酶组sirna文库中存在的相同的623种激酶。对于高流通量合成,用于每个sgrna转录的模板是对含有tracrrna序列的共同80聚体退火的基因特异性的60聚体寡核苷酸(46)。
对于筛选,从spcas9-敲入小鼠中免疫淘选rgc,并且在384孔板中以每孔1,000个细胞接种。用neuromag(ozbiosciences)以每孔30ng一式两份地逆转染sgrna文库。因为1nmlzksipool良好地工作,以使全基因组sirna文库对dlk抑制致敏,所以将相同的试剂加入该筛选的所有孔中。每块板将包含阴性(tracrrna)和阳性(dlk)对照sgrna,其充当质量控制和标准化标准(以允许板对板比较)。48小时后,每孔将用1μm秋水仙碱处理,以促进dlk/lkzàjnk信号传导并且降低本底存活(15,47)。更后,另外48小时后,使用celltiter-glo测量存活,并且针对每块板上的中值阴性对照孔标准化。基因基于三种sgrna的平均和中值活性进行评分且排序。活性sgrna在spcas9和野生型rgc中重新测试,以确保活性是spcas9依赖性的。与sirna一样,二次筛选通过合成其序列在初始筛选中未测试的新sgrna执行。
鉴于用rna转染原代rgc且测量存活的平台已进行广泛测试,未预期关于筛选本身的任何问题。此外,鉴于来自全基因组sirna筛选的结果,所述筛选使用lzksipool使系统对dlk抑制致敏并且产生dlksirna微型库作为16,877种基因中的**命中,该策略能够鉴定dlk途径成员。主要问题是合成sgrna文库中涉及的劳力。进一步考虑合并三种sgrna的模板和体外转录库。通过使所有插入缺失偶然地作为三个核苷酸的倍数结束,靶向相同基因的多重sgrna应该减少给定基因座逃脱靶向的机会。然而,至少对于lzk和dlk,未观察到使用合并策略增加的功效(图19j),可能是由于已经很高的插入缺失频率(图19h)。为了避免具有太少可接受的spcas9靶的一些较小激酶,sitools(munich,德国)用于测试具有与锤头状核酶的5'融合的sgrna(图23),从而消除了关于sgrna以鸟嘌呤开始的需要(因为t7转录将以核酶开始),并且将靶数目增加四倍。已显示该系统产生正确的切割产物,并且目前正在测试以这种形式制备的sgrna的功效(图23)。更后,需要时,小鼠sgrna文库现在从dharmacon商购可得。涉及基因特异性crrna和常见tracrrna的共转染的dharmacon仅具有上文使用的嵌合sgrna的50%活性(数据未显示)。如果成功,则下一步将是设计且筛选全基因组sgrna文库。
2.筛选sirna文库,分组至靶激酶网络,以便鉴定其敲低改善rgc存活的基因。
即使激酶在rgc细胞死亡中起关键作用,其在抑制时的表型也可以被冗余/补偿性激酶掩盖。致敏筛选是避开这种局限性的一种方法,但依赖已经知道的冗余对的一个成员(例如全基因组筛选中的lzk)。作为替代的更无偏的方法,可以筛选sirna文库,其中每个孔同时靶向多重激酶,因为它们可以是冗余信号传导网络的部分而分组。更初利用sigma激酶组sirna文库,浓缩成623个微型库(每种基因三个sirna),然后将微型库以新型组合进行组合。由于尝试所有成对激酶组合(6232)是不可行的,因此三种不同的生物信息学方法用于创建微型库的库。首先且在概念上更简单的,靶向具有更高同源性程度的激酶(例如jnk1、2和3)的sirna将被分组在一起。基于ensembl数据库,sigma激酶组sirna文库中的488种基因与另一个成员共享显著的同源性,导致1626个对(因为给定的基因可以与多于一个其他成员同源)。为了使数目更容易管理,这些对进一步聚类成111个组,具有每组四个成员的中值。其次,具有重叠底物谱的激酶可能是多余的。在由4,191种独特的全长人蛋白组成的蛋白质微阵列上执行了关于289种人激酶的磷酸化反应,并且鉴定了3,656种激酶-底物关系(48)。在分析中包括的sigma激酶组sirna文库中的198种基因中,存在具有其底物谱的显著重叠的2,089对。因为激酶可以具有与多于一种其他激酶的重叠谱,这导致3,386个潜在组,具有每组11种基因的中值。更后,利用所有121种激酶及其所有成对组合在酿酒酵母中进行遗传相互作用测定,以便产生全面的上位图(e-map)的事实(49)。推断这些成对遗传相互作用中的一些可能在小鼠中是保守的,并且在一些情况下,指示冗余/补偿途径成员(合成致死性相互作用的遗传底物)。在具有酵母同源物的sigma文库中的223种激酶中鉴定出1,674个成对激酶相互作用,产生具有两个基因的中值大小的1,003个组。总共有4,500个sirna组,其中更大的组含有19种激酶。更后,执行先导实验,以确保具有更多靶(例如19种激酶)(其中大多数可能是无活性的)的库不会稀释出活性对。令人欣慰的是,注意到用dlk和lzksirna微型库转染的rgc即使用无活性sirna稀释64倍也产生可检测的表型(图24a)。筛选本身将一式两份完成,其中每块板含有阴性(lzk/lzk)和阳性对照(dlk/lzk),用于协同作用和作为标准化标准。存活数据将使用我们已生成且挖掘的种子校正因子进行调整,以寻找在多个存活促进孔中出现的组合。然后解卷积这些库以鉴定活性介质。更后,二次确认涉及使用其序列未在sigma文库中测试的独立sirna。
我们已确认,我们生成激酶组合的三种方法产生的列表包括至少一种已知的相互作用激酶(例如磷酸网络-jnk1/2/3和mkk4/7;同源物-jnk1/2/3、mkk4/7和dlk/lzk;酵母e-map-dlk/lzk)。另外,因为筛选可以在几周内完成,所以该过程可以是迭代的,其中筛选的结果导致分组算法中的修改。鉴于对labcyteecho550声学液体处理器的接近,文库的实际构建应该相对容易,所述处理器可以在几小时内组装所有4,500个组,而无需使用移液器吸头。一个障碍可能是单个孔中如此多的sirna的组合可能导致众多脱靶相互作用。作为补充或替代策略,sitools(munich,德国)可以用于使用sipool构建小鼠激酶组文库。这具有使中靶效应富集30倍的优点。实际上,当用sirnas相对于靶向激酶的sipool转染a439细胞且测定存活时,靶向相同基因的sirna具有仅0.19的r2(图24b)。相比之下,靶向相同基因的独立sipool具有0.71的r2。
3.测定来自上文节段1或2的验证命中是否影响dlk/lzk信号传导。
与对于dlk和lzk完成的一样(图19e),rgc用靶向验证激酶的sirna、sipool或sgrna(来自上文节段1和2)单独或与lzksipool组合(以使细胞对途径抑制敏感)转染,并且在24-48小时,对于磷酸化的mkk4、mkk7(dlk/lzk的直接底物)、jnk和jun进行免疫印迹。其敲除或敲低压制jnk途径信号传导的激酶视为dlk/lzk的潜在上游激活物。促进存活而不影响mkk/jnk磷酸化的那些必须减少对dlk的敏感性,因为它足以触发rgc细胞死亡。
虽然sirna的效应易于在未选择的整个群体中检测到,但sgrna更困难,因为敲除不在的细胞中发生。为了解决这个问题,rgc用sgrna进行转染,并且经历用秋水仙碱的标准96小时方案,以便提供强大的选择压力且富集敲除细胞。然后,根据剩余细胞的数目,通过关于jnk磷酸化的免疫印迹或使用针对磷酸化-jun的抗体的免疫荧光来测量jnk途径状态。
4.测定来自节段1或2的验证命中是否在dlk和lzk的上游或下游遗传上起作用。
rgc如节段3中进行转染,然后在足够的选择时间后,用过表达野生型大鼠dlk或人lzk的腺病毒攻击剩余的细胞。dlk/lzk上游的基因应该具有由dlk/lzk过表达逆转的其敲低/敲除表型。作为互补方法,产生过表达关于候选激酶(包括致使激酶组成性激活的任何已知突变体)的cdna的腺病毒,并且测试其过表达是否促进rgc细胞死亡以及它是否可以用dlk/lzk抑制剂和/或dlk/lzksirna阻断(如将预期的,如果它们在dlk/lzk的上游起作用)。
可以执行用我们容易获得的野生型大鼠dlk表达腺病毒而不是真正的组成型活性突变体逆转存活表型。如果可以逆转存活表型,则亚克隆并表达dlk突变体(s584a/t659a),其被认为具有增加的活性(50)。更后,尽管上文节段1的主要目的是鉴定dlk和lzk的上游激活物,但追求看起来在dlk/lzk下游/独立于dlk/lzk起作用的激酶。
5.鉴定来自节段1或2的验证命中是否促进存活而不影响轴突再生。
rgc用靶向验证激酶的sirna、sipool或sgrna(来自节段1和2)单独或与lzksipool组合(以使细胞对途径抑制敏感)转染,并且在72小时后用钙黄绿素am染色。cellomics自动显微镜用于使细胞成像并且计算总体神经突长度。理想地,将鉴定其抑制促进存活而不影响神经突向外生长的基因。
为了证实神经突向外生长测定可能预测体内再生,我们先前已显示,与对照处理的细胞相比,用dlk/lzk抑制剂和/或sirna处理的原代rgc具有急剧减少的神经突长度(图24c)。此外,节段2中的互补实验可以用于验证我们发现的体内相关性。
6.开发与aav递送相容的crispr系统,以便在体内敲除rgc中的靶基因功能。
筛选工作先前已鉴定rgc中的多重神经保护(europrotective)靶,并且节段1和2中概述的方法将进一步扩展候选物列表。另外的步骤包括:在视神经病变的啮齿类动物模型中体内验证这些命中,验证命中的优先次序,以及优先靶的特定抑制剂的后续开发。潜在理想的方法是基于基因疗法的策略,其既可以快速验证命中(即,体内敲除基因而不必创建/获得敲除小鼠),并且以后又充当治疗剂本身。如图19g-j中证实的,基于crispr的系统可以用于rgc中,以在体外稳固地破坏基因功能。crispr技术将用于rgc的体内基因组编辑,利用aav2有效转导rgc的能力(9,10)。不幸的是,aav衣壳具有~4.8kb的包装容量,其已阻止在单一病毒中递送spcas9-表达盒(~4.8kb)和grna表达盒(~0.4kb)两者。aav/crispr系统已在体内使用,但不得不依赖:1)两种不同aav的使用(51),这对于基因治疗是亚更佳的,或者;2)较小的金黄色葡萄球菌cas9(sacas9)的使用(52),其具有更限制性的pam并且将潜在靶位点的数目减少四倍。这个问题由于以下事实而复杂化:grna通常脱离u6启动子而表达,所述u6启动子在鸟嘌呤处起始,将靶位点限制为gn19ngg(spcas9)或gn19nngrrt(sacas9)。更近,已显示可以在腺嘌呤或鸟嘌呤处起始的h1启动子使潜在的cas9靶数目倍增(53)。天然地,h1启动子在一个方向上驱动其经典poliii转录物h1rna的转录,并且在另一个方向上驱动称为parp2的遍在表达的polii转录物的转录(54,55)(图28a)。这种紧凑的(~0.2kb)双向h1体系结构允许spcas9和grna从单个启动子表达,并且包装到单个aav载体内。
为了测试该系统,生成了aav构建体,其使用h1启动子以在polii方向上表达mcherry-组蛋白2b融合物和在poliii方向上表达dlk#4grna(5’gnnnnnnnnnnnagatctnnngg3’(seqidno:163))(其方便地靶向bglii位点(图19g和1h))(dlkgrna:h1:h2b-mcherry)。因为总大小低于2.4kb,所以反向末端重复之一突变,以生成已知增加和加速基因表达的自互补的(sc)aav(56)。产生病毒颗粒并且用于转导从spcas9-p2a-gfp敲入小鼠分离的原代rgc。在陶扎色替的存在下仅五天后(以预防细胞死亡且避免选择),观察到具有接近转导的稳健报道基因表达(图25b,左)和bglii位点的大于90%丧失,即基因编辑(图25b,右)。分开地,用由单个h1启动子表达spcas9和grna两者的质粒(非病毒)转染nih3t3细胞,并且显示它导致稳健切割,与传统的双载体系统可比较(图25c)。总之,这些研究验证了scaav系统(待在节段8、9和15中使用)和双向h1启动子的polii/iii能力。如下所述,生成了表达spcas9和grna两者的单一aav递送系统。在一些实施方案中,关于自互补的(sc)aav的序列可以包含选自seqidno:1398-1400的核苷酸序列。
为了帮助转导后的rgc分析,开发了基于流式细胞术的方法用于定量和表征小鼠rgc。将视网膜分离,解离且用丙酮固定(以保存核酸品质),然后用针对rgc抗原γ-突触核蛋白(sncg)和β-iii-微管蛋白(tuj1)的抗体染色。sncg+/tuj1+双阳性细胞代表rgc,如通过以下发现证实的:它们还就另外的rgc标记物thy1.2和neun染色(图26a),它们的存活在视神经挤压后减少(图26b和25c),并且纯化的原代rgc具有相似的概况(图26b)。然而,与thy1.2和neun不同,sncg和tuj1的表达不被轴突损伤下调(数据未显示),并且因此它们在视神经病变模型中仍然是有用的标记物。更后,该方法与细胞分选相容,并且显示分选的细胞具有与免疫淘选的rgc非常相似的蛋白质组学概况和完整的rna/dna(数据未显示)。
7.敲除dlk作为基于scaav/crispr的方法可以用于在spcas9-敲入小鼠体内靶向基因的原理证明。
使用我们已在体外验证的病毒(图25b),我们将优化体内病毒滴度。表达spcas9-2a-gfp的小鼠接受以1010、109、108或107个病毒颗粒/μl的病毒的单侧玻璃体内注射(~1μl)(每组四只小鼠)。两周后,对于aav生命周期完成给出足够的时间,rgc如上所述进行定量且分选。
更后,纯化基因组dna且测定dlk破坏,如通过bglii消化的丧失测量的,以便测定更佳滴度(即更大量的切割而不引起rgc毒性)。在更后一步中,表达spcas9的小鼠在一只眼中用更佳剂量的scaav2-dlkgrna:h1:h2b-mcherry或表达非靶向grna代替dlkgrna的对照病毒进行玻璃体内注射。两周后,眼将经历三秒视神经挤压(以便不损害血管)或假对照手术,并且在另外两周后,通过sncg/tuj1流式细胞术测量四组中的存活(n=10个视网膜/组)。将来,这个系统是否相容将通过aav9载体的静脉内施用进行测试,因为该方法已显示有效转导rgc(57,58)。
基于已描述的floxeddlk小鼠实验,已知dlk的靶向破坏改善了rgc存活(5)。此外,已知基于crispr的敲除在体外起作用。因此,看起来在用dlkgrna转导的那些rgc中检测到存活中的增加是高度可能的。如果未看到>50%的切割或存活表型,则分析表达mcherry的aav转导的细胞。这应该是容易实现的,因为流式细胞术技术目前可以与多达四个通道一起工作(图26a)。
8.使用scaav/crispr系统以验证sa1中鉴定的命中是小鼠视神经挤压模型中rgc细胞死亡的介质。
其敲低或敲除改善体外rgc存活的经鉴定的基因将在体外用grna靶向,并且如节段7中对于dlk描述的进行测试。优先考虑看起来在dlk上游起作用或似乎不影响再生的那些基因。与对于dlk完成的,关于每种靶基因的多重grna将在原代rgc中进行测试,以鉴定前进到aav系统内的更有效grna。更后,通过对于磷酸化的jun和dlk免疫染色视网膜铺片来验证假设在dlk上游的靶,以测定它们是否体内压制途径(5)。
由于筛选鉴定了活性grna,所以起于节段1的基因应该直接在体内进行测试。然而,因为筛选用lzksipool致敏,所以可想象体内表型需要lzk或dlk抑制。幸运的是,当制备lzk条件敲除时,使用首先产生无效等位基因的策略。此外,与dlk-null小鼠的情况不同(59),lzk-null小鼠是活的且能育的。因此,将lzk-null小鼠与spcas9-敲入系杂交以产生rosa26cas9/cas9lzk-/-小鼠,确认节段1命中的更适当系。
节段2中鉴定的基因在体内验证可能更具挑战性,因为它们可能需要同时破坏多重基因。对于那些,利用构建体的小尺寸,以及可以容纳另外的h1驱动的另一种grna表达并且仍维持相同的scaav设计的事实。这允许用一种病毒载体敲除两种基因(在节段11中更多地讨论)。未来可以尝试使用多于两个grna(代替mcherry或在单链(ss)aav设计中),以便靶向基因网络。
9.使用修改的scaav2/crispr方法,以验证节段5中鉴定的命中不影响再生。
视神经挤压后rgc再生的基础水平极低,但可以通过pten的抑制得到增强,尽管pten和dlk同时被抑制时不是如此(6)。作为测试节段5中鉴定的候选物的抑制是否促进rgc存活而不影响再生的一种方法(与dlk不同),获得关于每种候选基因的敲除小鼠并且将其在ptenf1/fl背景下繁殖-这需要巨大量的时间和工作。相反,体内敲除方法用于生成scaav,其在poliii方向上从标准双向h1启动子表达靶向目的基因的grna,并且在polii方向上表达无h2b融合的mcherry(以便显现轴突)。第二个h1启动子将表达ptengrna。表达spcas9的小鼠在一只眼中用更佳剂量的scaav2-xgrna:h1:mcherry;h1:ptengrna(其中x是非靶向grna)(用于预防再生的阴性对照)、dlk#4grna(用于预防再生的阳性对照)、或靶向来自节段5的候选物的grna进行玻璃体内注射。两周后,眼将经历三秒视神经挤压(以便不损害血管)。更后,在另外两周后,将收获来自三组的视神经(n=10个视神经/组),纵向切片并且定量延伸超出挤压部位的mcherry阳性轴突的数目。
如果pten缺失的再生表型不足,则可以加强对socs3的效应(33,35,60)。提出这些实验作为替代方案,因为关于dlk在再生中的作用的开创性工作使用pten(而非组合的pten/socs3)缺失(6)。多等位基因破坏是费劲的,并且可能以低频率发生。幸运的是,即使有两个h1启动子和scaav设计,紧凑的h1启动子留下了接近1kb的包装空间,这允许在基于荧光蛋白的报道基因中改造crispr编辑(61)。
10.敲除dlk作为治疗上合适的基于ssaav/crispr的方法可以用于靶向野生型小鼠中的基因的原理证明。
节段7和8中概述的实验利用了scaav的稳健性,并且允许生物学的快速验证,但由于它们依赖spcas9敲入小鼠而在治疗上无用。作为互补方法,h1启动子的双向特点用于驱动cas9和grna的表达,允许两个盒均被包装到单个病毒载体内。尽管该策略允许在大小方面与野生型aav可比较的插入片段,但它对于scaav太大了。因此,ssaav2-dlkgrna:h1:spcas9颗粒如节段7中所述进行制备且测试,除了动物不表达spcas9(因为它现在由病毒提供)之外。该病毒将在青光眼模型中进行测试,并且包括视力的功能测量。重要的是,虽然dlk和rgc一直是焦点,但这些数据将具有甚至更深远的前述-使用单一aav/crispr病毒以修饰基因组并增加对疾病状态的抗性。
经历双等位基因破坏的细胞的百分比可能不足以可靠地检测视神经挤压模型中的存活增加。迄今为止,已使用体外模型对接近20个dlk靶位点进行取样,并且未发现比#4(用于节段6)工作好得多的位点。甚至dlk的单等位基因破坏也可以增加存活,尽管低于由双等位基因破坏赋予的稳健保护(37)。为了使细胞对这种单等位基因dlk破坏致敏,可以使用lzk-null小鼠重复实验。互补策略是延长视神经挤压与rgc定量之间的时间。尽管,实验通常在两周时停止(当~75-80%的rgc已死亡时),接下来的1-2周发生进行中的丧失,更终达到~90-95%细胞死亡的平台期。即使仅5%的细胞具有dlk的双等位基因破坏,这也可以产生在一个月时存活细胞的数目中的50-相对增加(与两周时的20-25%增加相比)。更后,尽管dlk#4靶位点是优选的(由于bglii位点),但可以用#5重复实验,所述#5在小鼠和人中具有确切相同的序列,并且因此允许测试人治疗剂的动物模型。
11.测量通过节段10中开发的aav/crispr治疗剂产生的脱靶。充分确定spcas9在pam远端的前间区序列部分中更好地耐受错配(62,63),因此更可能的脱靶具有保守的3'序列和在5'末端中的1-2个错配。
因为bglii位点定位在切割位点附近(3'),所以关于#4grna的几乎所有可能的脱靶都保留了bglii位点。可以使用节段10中获得的基因组dna,并且可以使用图19h中描述的bglii测定,以定量在更可能的脱靶位点处的切割。
虽然体外特异性(图19h)是令人鼓舞的,但脱靶切割可能是一个问题。如果是这种情况,则两组更近描述的突变之一(spcas9-hf1-n497a、r661a、q695a、q926a;espcas9-k810ak103a、r1060a)可以用于spcas9中,其改善特异性(64,65)。替代方法将是测试另一种grna序列,尽管测量中靶和脱靶切割必须用更费力的方法,如t7核酸内切酶测定和测序来完成。这种想法的变体是从#4grna的5'末端截掉1-3个核苷酸,这是已显示改善特异性的技术(66)。幸运的是,#4grna以gaag开始,因此这些截短中的任一种仍与h1启动子(其在a或g处起始)相容。更后,值得一提的是,h1表达的grna可能产生较低的脱靶切割率(53)。
12.开发aav/crispr治疗剂,其在单一载体中递送cas9和两个grna。
通过使用h1启动子,dlkgrna:h1:spcas9具有4.5kb的大小,远低于野生型病毒的4.7-4.8kb大小和5.2kbaav包装限制。这提供了用另外的h1-grna盒(~0.2kb)修饰节段10中提出的构建体的机会。由单一病毒表达两个grna(除spcas9之外)开放了几种可能性,包括使用更特异性的“切口酶”突变体spcas9(67),在单个基因中生成大缺失(与我们工作的主题有关),和同时靶向两个补偿基因。为了测试这一点,将h1:lzkgrna#1加入表达盒中至节段10中描述的构建体,并且生成ssaav2-dlkgrna4:h1:spcas9;h1:lzkgrna5病毒颗粒。这些如节段10中所述进行测试,这次比较靶向单独的或与lzk组合的dlk的病毒(n=10个视网膜/组)。
该构建体的大小推动了aav包装容量的限制(尽管仍然大于野生型病毒小于0.1kb)。如果这变成问题,则可以使用较小的小鼠h1启动子(其也可以双向起作用)或sacas9,其比spcas9小接近1kb(52)。后一种方法需要不同的靶位点,因为#4和#5位点都不与sacas9pam要求相容。
13.探索dlk通过其调节mef2a以促进rgc细胞死亡的机制。
mef2a在肌肉分化和心血管生理学中具有充分研究的作用(68)。在神经元中,mef2a已显示在促进存活方面起关键作用(27),并且尽管mef2a的脑特异性条件性敲除不影响总体神经元存活,但mef2a/c/d的组合敲除显示神经元细胞凋亡中的明确增加,导致早期产后致死率(30)。响应兴奋性毒性刺激,皮质神经元利用两种机制以抑制mef2a信号传导并促进细胞凋亡:半胱天冬酶催化mef2a的切割,其导致mef2a的显性失活活性(69)和其s408的细胞周期蛋白依赖性激酶5(cdk5)磷酸化,这导致反式激活结构域(70)的失活,或者,尤其是在树突/突触生理学的情况下,在k403处的后续类泛素化(sumoylation)和转录阻遏形式的mef2a的形成(29,71)。在这些情况的每一种中,mef2a促进神经元存活,并且mef2a的抑制导致细胞死亡。相比之下,mef2a被鉴定为体外和体内rgc细胞死亡的介质(图20f和20g)。当将floxedmef2argc的存活与floxedmef2a/c/drgc进行比较时(30),没有观察到差异(数据未显示),提示mef2a不干扰mef2c/d以促进rgc细胞死亡。更后,尽管响应体内视神经损伤或体外免疫淘选,检测到稳健的dlk依赖性mef2as408磷酸化,但不可磷酸化的s408a突变体不干扰活性(图20i)。总之,这些结果提示dlk激活导致新型mef2a改变,其负责细胞死亡。
14.使用蛋白质组学方法,以鉴定mef2a中的dlk依赖性翻译后修饰。
为了鉴定dlk/lzk通过其调节mef2a的机制,mef2a从floxeddlk/lzk原代rgc中纯化,所述原代rgc用adeno-cre(以便取消dlk/lzk信号传导)、adeno-gfp(其具有活性dlk/lzk信号传导)、野生型大鼠dlk(其超激活途径)或激酶死亡大鼠dlk(其作为显性失活起作用)进行转导。在这些位点各自处的磷酸化残基和磷酸化的相对丰度将通过在我们团队可用的几种orbitrap质谱仪(thermoscientific)之一上的串联质量标签(tmt,thermoscientific)定量以及液相层析串联质谱法(lc-ms/ms)分析进行测定。这种方法还允许分析其他翻译后修饰,如类泛素化和乙酰化,已经知道其在mef2a信号传导中起作用(29,71)。看起来取决于dlk/lzk的存在的变化,且尤其是随着途径的超激活而增加的那些变化将在节段15中进行跟踪观察。
鉴于mef2a翻译后修饰将在我们疾病相关的原代细胞系统的背景下进行测定,更大的挑战可能是原材料的量。为了避开该问题,可以常规分离3-5,000,000个rgc,鉴于ip的低复杂性(即仅察看一种蛋白质),这应该是绰绰有余的。如果不能获得足够的mef2a蛋白,则可以使用加上表位标签的mef2a的过表达。其次,在**优化用于这些物种的mrm测定后–经由靶向潜在mef2a磷酸化位点(已知和未知的两者)的多重反应监测(mrm)方法,可以增加ms分析的灵敏度和准确度(72,73)。如果在多重同时位点处的磷酸化对于通过dlk/lzk的mef2a调节是关键的,则可以对完整的非胰蛋白酶消化的mef2a采用自上而下的蛋白质组学方法,以确定磷酸化的多重性。
15.使用突变和功能获得/功能丧失实验的组合,以验证蛋白质组学鉴定的翻译后修饰。
节段14中鉴定的翻译后修改将在原代rgc系统中进行测试。将关于每个残基的突变体(例如s408a)改造到小鼠mef2acdna内,并且如上文多次描述的穿梭到腺病毒载体内。然后将rgc从floxedmef2a小鼠中分离,并且用腺病毒-cre(moi1000)转导,以促进存活并消除内源性mef2a。48小时后,腺病毒将用于重新引入野生型mef2a或不可修饰的突变体。如果翻译后修饰对于mef2a依赖性杀死是必需的,则该突变体预期是功能丧失。此外,野生型dlk的过表达(其超激活途径且加速rgc细胞死亡)将与突变体组合使用,以察看它们是否具有显性失活活性(即,如预期的,该修饰是否是dlk信号传导通过其激活mef2a的机制的部分)。更后,至少在磷酸化位点(phosphosites)的情况下,改造磷酸化模拟突变(例如s408e),且测试即使在组合的dlk/lzk抑制(使用sirna)的背景下,这些是否获得组成型活性并促进细胞死亡。
16.使用crispr/aav策略,以测试体内mef2a的靶向破坏是否促进rgc存活而不影响轴突再生。
尽管dlk在细胞死亡和轴突再生两者中起关键作用,但鉴定了介导细胞死亡信号的四种下游转录因子。虽然其中至少两个(即sox11和jun)在再生中具有已知的作用,但mef2a的作用未知。因此,mef2a可能潜在代表神经保护靶,其解离存活和再生。实际上,在体外,mef2a敲低似乎不严重影响神经突向外生长(数据未显示)。通常,这种研究方法具有有限的效用,因为作为转录因子的mef2a不易于药物靶向。然而,节段10中概述的策略允许我们治疗上靶向基因如mef2a。此外,节段9中描述的方法(其中多重基因被敲除,并且甚至选择性地标记具有活性crispr编辑的细胞)提供在体内容易地测试mef2a生物学的机会。因此,spcas9-2a-gfp敲入小鼠将用节段9中描述的病毒进行转导,修饰以靶向mef2a、dlk(用于预防再生的阳性对照)或非靶向对照(用于预防再生的阴性对照),并且如节段9中描述的进行测定。
结论
总之,本文描述的是旨在进一步探索dlk和lzk通过其促进rgc细胞死亡的上游和下游机制的一组实验。此外,使用crispr/aav治疗剂,已开发了平台,其允许体内快速验证,并且更重要的是,无缝过渡到基于高度特异性的基因疗法的治疗剂。实际上,虽然存在关于基于crispr的治疗剂用于基因组编辑的巨大兴奋,但该领域受到spcas9和grna不能同时包装到单个病毒载体内的事实的限制。使用h1启动子,已克服了这个障碍。使用dlk作为实例,基因组修饰可以在治疗上用于视神经病变中。
靶:
dlk
外显子1:
lzk
外显子1:
实施例5
方案
将hek293细胞接种在t25烧瓶中,并且在补充有10%胎牛血清和抗生素的dmem中生长至半汇合。铺平板两天后,用质粒paav-cep290转染一个烧瓶中的细胞。向细胞培养基中加入在总共500ul无添加剂的optimem培养基中含有4ug质粒dna、20ullipofectamine3000(thermofisher/invitrogen)和10ulp3000试剂(thermofisher/invitrogen)的转染混合物。用100ulaav2-cep290(2.06e10^11个病毒基因组)的包装和纯化的原液感染第二个烧瓶中的细胞。第三个t25烧瓶是未处理的且充当对照。转染或感染后48小时,在胰蛋白酶-edta中收获细胞,并且通过离心沉淀。对于每个样品,将细胞沉淀重悬浮于200ulpbs中,并且按照制造商的方案用qiagendnamini试剂盒进行加工。将基因组dna在200ul水中洗脱。
对于t7endoi分析,通过在quickextract溶液(epicentre,madison,wi)中重悬浮细胞,在65℃下温育20分钟,然后在98℃下温育20分钟,来提取基因组dna。提取溶液直接使用或使用dnacleanandconcentrator(zymoresearch,irvine,ca)清洁,并且通过nanodrop(thermo30fisherscientific)定量。使用phusiondna聚合酶(newenglandbiolabs),从~100ng基因组dna扩增crispr靶位点周围的基因组区域。合并多重独立的pcr反应物,并且使用dnacleanandconcentrator(zymoresearch,irvine,ca)纯化。使在10mmtris-hcl、50mmnacl、10mmmgcl2和100μg/mlbsa中含有150ngpcr产物的25μl体积变性,并且缓慢再退火以允许形成异源双链体:95℃10分钟,95℃至85℃以-1.0℃/秒渐变,85℃1秒,85℃至75℃以-1.0℃/秒渐变,75℃1秒,75℃至65℃以-1.0℃/秒渐变5,65℃1秒,65℃至55℃以-1.0℃/秒渐变,55℃1秒,55℃至45℃以-1.0℃/秒渐变,45℃1秒,45℃至35℃以-1.0℃/秒渐变,35℃1秒,35℃至25℃以-1.0℃/秒渐变,然后保持在4℃下。向每个反应中加入1μlt7endoi(newenglandbiolabs),在37℃下温育30分钟,然后立即置于冰上。对于凝胶分析,将3μl反应物与3μl2x上样染料(newenglandbiolabs)混合,上样到6%tbe-page凝胶上,并且在显现之前用sybrgold(1:10,000)染色~15分钟。凝胶在gellogic200imagingsystem(kodak,15rochester,ny)上显现,并且使用imagejv.1.46定量。使用二项式导出的方程计算nhej频率:
%基因修饰=100*(1-(sqrt(1-((a+b)/(a+b+c)))))
其中“a”和“b”的值等于在本底扣除后的切割片段的积分面积,并且“c”等于在本底扣除后未切割的pcr产物的积分面积。
对于限制性分析,通过在quickextract溶液(epicentre,madison,wi)中重悬浮细胞,在65℃下温育20分钟,然后在98℃下温育20分钟,来提取基因组dna。提取溶液直接使用或使用dnacleanandconcentrator(zymoresearch,irvine,ca)清洁,并且通过nanodrop(thermo30fisherscientific)定量。使用phusiondna聚合酶(newenglandbiolabs),从~100ng基因组dna扩增crispr靶位点周围的基因组区域。合并多重独立的pcr反应物,并且使用dnacleanandconcentrator(zymoresearch,irvine,ca)纯化。使在10mmtris-hcl、50mmnacl、10mmmgcl2和100μg/mlbsa中含有150ngpcr产物的25μl体积在37℃下消化1小时。对于凝胶分析,将3μl反应物与3μl2x上样染料(newenglandbiolabs)混合,上样到6%tbe-page凝胶上,并且在显现之前用sybrgold(1:10,000)染色~15分钟。凝胶在gellogic200imagingsystem(kodak,15rochester,ny)上显现,并且使用imagejv.1.46定量。使用方程计算nhej频率:
%基因修饰=((a+b)/(a+b+c))*100
其中“a”和“b”的值等于在本底扣除后的切割片段的积分面积,并且“c”等于在本底扣除后未切割的pcr产物的积分面积。
利伯氏先天性黑蒙(lca)由一组早发型儿童视网膜营养不良构成,其特征在于在生命的前几个月期间严重的视网膜功能障碍和严重的视力损害或失明。lca,作为罕见疾病,是遗传性失明的更常见原因,构成所有已知遗传性视网膜变性疾病的多达5%。关于lca10–对于其不存在fda批准的治疗的一种疾病–的更常见原因是cep290基因中的深度内含子突变(denhollander,a.i.等人(2006)americanjournalofhumangenetics79,556-561)(图51)。ivs26c.2991+1655a>g突变导致从头的强剪接供体位点,以及隐藏外显子(外显子x)后续包括在cep290mrna内。cep290,位于光感受器的连接纤毛中的中心体蛋白,在纤毛发生和纤毛运输中起作用(drivas,t.g.等人(2013)thejournalofclinicalinvestigation123,45254539;craige,b.等人(2010)thejournalofcellbiology190,927-940;tsang,w.y.等人(2008)developmentalcell15,187-197)。包括含有过早终止密码子的外显子x导致截短形式的cep290和更终的光感受器变性。
一些形式的lca潜在顺应通过重组腺伴随病毒(aav)的治疗,所述重组腺伴随病毒(aav)被改造为递送缺陷型细胞基因的功能性拷贝。在2008年,在i期临床试验中,互补rpe65中的突变的转基因通过aav成功地递送至lca2患者(maguire,a.m.等人(2008)thenewenglandjournalofmedicine358,2240-2248)。注意到一些应答,但不幸的是,效应并不持久,潜在地由于丧失转基因表达(azvolinsky,a.(2015)natbiotechnol33,678;schimmer,j.等人(2015)humangenetherapy.clinicaldevelopment26,208-210)。此外,导致不同lca亚型的一些基因(如cep290)中的突变对于aav递送而言只是太大,并且因此通过基因治疗方法仍然无法治愈。此外,光感受器对蛋白质水平高度敏感(olsson,j.e.等人(1992)neuron9,815-830;tan,e.等人(2001)investigativeophthalmology&visualscience42,589-600;seo,s.等人(2013)investigativeophthalmology&visualscience54),并且转基因实验已证实来自cep290过表达的光感受器毒性(burnight,e.r.等人(2014)genetherapy21,662-672)。鉴于下述特性,我们认为cep290突变作为用于crispr-cas9技术的治疗靶特别有吸引力。
crispr-cas9技术的发展已彻底改革了基因编辑领域,并且提供了治疗遗传疾病的全新方法。crispr-cas9系统由指导rna(grna)构成,所述grna以序列特异性形式靶向cas9核酸酶。通过crispr系统的切割需要grna与dna序列的互补碱基配对和必需的前间区序列-相邻基序(pam),在靶位点3'发现的短核苷酸基序(doudna,j.a.等人(2014)science346,1258096;hsu,p.d.等人(2014)cell157,1262-1278)。目前,限制性更小且更常用的cas9蛋白来自化脓性链球菌,其识别序列ngg,并且因此,crispr靶向序列是n20ngg。虽然众多研究已显示疾病突变可以在体外有效靶向,但用于体内使用的基于crispr-cas9的治疗剂的开发受到安全性问题和递送约束的阻碍。
虽然疾病突变的crispr靶向已显示在众多体外背景下以及通过小鼠和其他动物研究在体内有效,但所有目前方法离临床使用仍很遥远,这在很大程度上是由于递送约束。aav载体是眼基因治疗注射中更频繁和成功使用的病毒载体(dalkara,d.等人(2014)comptesrendusbiologies337,185-192;day,t.p.等人(2014)advancesinexperimentalmedicineandbiology801,687-693;willett,k.等人(2013)frontiersinimmunology4,261;dinculescu,a.等人(2005)humangenetherapy16,649-663)。一些特点使得aav成为更具吸引力的选择:病毒是非致病性的,它感染分裂细胞和非分裂细胞两者,表达可以持续很长一段时间,并且特别值得注意的是它在临床试验中的安全性、功效和毒性的一般缺乏的历史。另外,特定的aav血清型在视网膜下注射后靶向感光细胞方面是有效的。虽然aav载体提供了递送治疗性crispr组件的安全手段,但存在限制其效用的一个主要技术障碍–其大小。野生型aav基因组为长度~4.7kb,并且重组病毒可以包装至多~5.0kb(dong,b.等人(2010)moleculartherapy:thejournaloftheamericansocietyofgenetherapy18,87-92;wu,z.等人(2010)moleculartherapy:thejournaloftheamericansocietyofgenetherapy18,80-86)。该包装容量限定了可以用于单个病毒载体的dna的上限。
通过常规方法表达cas9和grna所需的dna超过5.2kb:polii启动子(~0.5kb)、spcas9(~4.1kb)、polii终止子(~0.2kb)、u6启动子(~0.3kb)和grna(~0.1kb)。aav递送攻击的一种方法是双载体方法:一种aav载体用于递送cas9,且另一种aav载体用于递送grna(swiech,l.等人(2015)natbiotechnol33,102-106)。然而,双aav方法利用小的小鼠mecp2启动子,一种已发现在视网膜细胞中表达的基因-除了视杆的关键例外(song,c.等人(2014)epigenetics&chromatin7,17;jain,d.等人(2010)pediatricneurology43,35-40)-提示除了由于病毒递送载量增加的潜在毒性外,共递送方法可能未能先验靶向绝大多数lca突变。虽然这是其他基因治疗介导的基因组编辑的潜在可行方法,但我们相反提议用于视网膜基因编辑的单一载体方法,其应增加效率,特异性靶向光感受器,并且减少来自病毒载量递送的潜在毒性。
我们更近报道,使用h1启动子而不是更传统使用的u6启动子来指导grna转录允许可用crispr基因靶向空间的大约倍增(ranganathan,v.等人(2014)naturecommunications5,4516)。值得注意的是,我们还检测到关于脱靶切割的较低倾向,这提示h1启动子对于治疗方法更有利。在这些研究期间,我们注意到与内源性h1rna基因的紧密基因组接近的蛋白质编码基因(parp-2)的存在(myslinski,e.等人(2001)nucleicacidsres29,2502-2509;baer,m.等人(1990)nucleicacidsres18,97-103)。h1rna(poliiirna转录物)和parp-2基因(polii转录物)的起点之间的序列是230bp(图52),指示这个相对较小的序列可以充当紧凑的双向启动子-据我们所知,这是哺乳动物基因组中可以指导polii和poliii转录物两者的**双向启动子序列。我们假设h1启动子的这些偶然性质可以允许我们克服将两种crispr组件包装到单个aav内的尺寸障碍。
我们将基于正交cas9系统并行开发两种crispr/aav治疗剂。首先是通过经由单一aav载体共递送spcas9和指导rna介导的lca10治疗剂的开发。其次是经由单一aav载体共递送sacas9切口酶和四种指导rna介导的lca10治疗剂的开发。这两种策略均对于更终临床使用而开发,并且因此安全性至关重要。更后,我们将生成含有lca10的同基因人干细胞系,用于表征和开发新型治疗剂。
通过单一aav载体共递送化脓性链球菌cas9(spcas9)和指导rna介导的lca10治疗剂的开发。
背景和原理。尽管众多研究已显示疾病突变可以在体外有效靶向,但用于体内使用的基于spcas9的治疗剂的开发已受到递送约束的阻碍。使用紧凑的双向启动子系统,我们已证实了通过单一aav载体共递送spcas9和grna的临床可行平台。spcas9提供了几个优点:它是更常用、更通用且更易理解的crispr系统。它的pam要求(ngg)比其他cas9蛋白明显更不严格,这依次又意味着可以直接靶向更多基因和更多突变。对于临床治疗方法重要的是,spcas9的蛋白质工程的更近进展已开发了多重高特异性/高保真变体,其具有低至零可检测的全基因组脱靶效应(kleinstiver,b.p.等人(2016)naturedoi:10.1038/nature16526;slaymaker,i.m.等人(2016)science351,84-88)。实际上,与其他cas9直向同源物靶向位点不同,内含子cep290突变落入spcas9位点内(图53),这使得lca10突变成为用于spcas9的特别有吸引力的靶。
通过定制grna序列,我们可以将spcas9(或spcas9变体)导向cep290突变,引起在剪接供体位点附近的双链dna断裂。对dna断裂的细胞应答主要通过两种竞争途径之一发生:非同源末端连接(nhej)或同源性定向修复(hdr)。nhej,用于dna修复的更主导途径,是易错途径,其导致在断裂点附近的缺失和插入,通常为+/-~15nt(mali,p.等人(2013)science339,823-826)。因此,通过使用细胞正常dna修复机制,我们可以破坏在剪接供体位点附近的序列,预防外显子x的包括,并且恢复正常的cep290剪接和功能。重要的是,预期该内含子区域中的许多突变在正常剪接中恢复。
我们的方法将涉及修改且优化crispr-cas9方法,使得所有需要的组件都可以通过单一aav5递送给光感受器,所述aav5是在哺乳动物杆视中具有记录性能的aav血清型。通过使用我们的h1-aav系统将cas9换成gfp,我们已能够证实使用aav5将gfp有效递送到光感受器(图2)。因为lca10是隐性遗传的,所以正常cep290剪接的50%拯救应该是足够的光感受器功能。
实验设计与方法。
1.grna的计算选择和分析。因为目标是更终临床应用的构建体正在开发中,所以必须小心地监测它们的潜在脱靶活性(wu,x.等人(2014)quantitativebiology2,59-70)。为此,我们将追求几种互补的方法。采用生物信息学方法,我们使用定制的perl脚本测定人基因组中所有潜在的crispr位点,所述定制的perl脚本编写为搜索spcas9靶向位点的两条链和重叠存在;例如,在过滤掉重复序列后,在人基因组中存在137,409,562个crispr位点。我们已使用bowtie(langmead,b.等人(2009)genomebiology10,r25)计算测定关于每个位点显示出脱靶效应的倾向,以将每个crispr位点重新比对回基因组上,允许在靶向序列自始至终至多3个碱基错配。我们的lca10spcas9位点分析鉴定了13个潜在的脱靶基因座,我们将测试所述基因座的假靶向:1个位点有2个错配,并且12个位点有3个错配(计算数据可在http://crispr.technology处获得)。侧接中靶和预测的脱靶位点的pcr引物将与高保真聚合酶(neb,phusion)一起使用,以扩增基因组序列,其然后通过t7ei测定进行测试。这允许我们在体外和体内监测我们的优化实验的靶向准确性。
2.人细胞系的体外评估。我们已开发了几种spcas9靶向质粒,其含有用于简单靶grna插入的独特限制性位点,以及侧翼noti位点,以允许容易地亚克隆到aav产生所需的含有itr的载体内。另外,我们将生成含有两个更近报道的高保真cas9变体spcas9-hf和espcas926,27的构建体(图54)。我们不知道这些变体之间的任何比较,并且使脱靶诱变更小化对于开发安全治疗剂是至关重要的。为了确保我们在每个位点处都具有有效的靶向,我们首先测定spcas9靶位点在hek293细胞中的切割。使用侧接靶位点的pcr引物,我们使用高保真聚合酶(neb,phusion)来扩增基因组序列,其然后通过t7ei测定(详细方案在http://crispr.technology处可获得)测试切割活性。
3.对于中靶/脱靶诱变的高流通量测序。我们旨在执行中靶和脱靶位点的位点特异性深度测序分析。使用高保真聚合酶(neb,phusion)将侧接crispr靶位点和预测的脱靶位点的基因组序列扩增15个循环,然后纯化(zymo,dnaclean&concentrator-5)。将纯化的pcr产物扩增5个循环,以附着illuminap5衔接子和样品特异性条形码,再次纯化,然后通过sybr绿色荧光定量,在bioanalyzer上分析,并且更后在用miseqpersonalsequencer测序之前,以等摩尔比合并。为了分析测序数据,使用illuminamiseqreporter软件对300bp配对末端miseq读数进行解复用,随后为原始读数的衔接子和质量修整。在所有读数上对野生型序列执行比对,并且nhej频率通过以下进行计算:100*(插入缺失读数的数目/插入缺失读数的数目+wt读数的数目)。
4.aav病毒生产。高滴度gmp样临床前aav5载体由我们的合作者(dr.williamhauswirth,universityofflorida)在他们的独立载体生产设施中,使用由他们实验室开发的无辅助质粒转染方法生成。通过碘克沙醇梯度离心,随后为q柱fplc层析来纯化载体,并且建立载体aav载体原种的glp样纯度,每个载体经受标准化组的物理和生物测定,包括纯度、生物负载、无菌性、含有dna的颗粒滴度、感染滴度、颗粒/感染率比和通过复制感受态aav的潜在污染的评价,每个都是临床试验indcmc部分的关键要素。
通过经由单一aav载体共递送金黄色葡萄球菌cas9(sacas9)切口酶和四种指导rna介导的lca10治疗剂的开发。
背景和原理。更近出现的通过aav递送crispr的另一种有希望的方法是使用较小的直向同源cas9蛋白。
由金黄色葡萄球菌cas9(sacas9)突出显示,所述sacas9由~3.2kb转录物编码(ran,f.a.等人(2015)nature520,186-191)。然而,使用sacas9的一个局限性是由于其pam要求(nngrrt)。由于pam序列越长,独特的基因组靶向位点的数目是spcas9的~1/4,并且不存在落在lca10a>g突变上的sacas9位点。特定突变不能被spcas9靶向(如上所述),因此我们的方法是采用缺失策略来去除隐藏外显子周围的小区域。sacas9基因的紧凑大小允许使用标准启动子和终止子元件,将其连同一个、两个或可能三个grna盒一起包装到单个aav载体内32。就基于crispr的治疗剂的安全性问题而言,更重要的无疑是脱靶诱变;如果cas9在非预期的位置处切割dna,则这可能发生。幸运的是,通过采用cas9的点突变(称为切口酶,其仅切割一条dna链),这种风险可以降低几个数量级。通过分别接合两个grna以在相反链上生成两个紧密相对的切口,cas9切口酶方法可以有效地生成双链断裂(图55)。如果两个grna识别在基因组中其他地方紧密接近且在相反链上存在的脱靶,则脱靶效应才可能发生,这种情况是统计上非常不可能的。出于这个原因,使用切口酶生成dna断裂是更安全的方法。关于生成缺失,该方法需要四个grna(每侧上一对以缺失靶向突变)33。然而,使用提供另外空间的h1双向系统,我们可以容易地容纳四个grna。
1.grna的计算选择和构建体生成。与我们上述生物信息学类似,我们已鉴定了基因组中的每一个sacas9靶向位点,并且将信息建立数据库(数据可在http://crispr.technology处获得)。为了使用sacas9切口酶系统靶向缺失,我们从我们的计算分析中鉴定了四个候选grna位点,其具有使用切口酶用于生成缺失的有利特性(friedland,a.e.等人(2015)genomebiology16,257;mali,p.等人(2013)natbiotechnol,doi:10.1038/nbt.2675;ran,f.a.等人(2013)cell,doi:10.1016/j.cell.2013.08.021)(图4)。
2.人细胞系中的体外评估。我们已构建了含有侧翼noti位点的sacas9靶向质粒,以允许容易地亚克隆到aav产生所需的含有itr的载体内。如同spcas9靶向质粒,这些质粒含有用于快速克隆特定grna序列的独特限制性位点。为了确保我们在每个位点处都具有有效的靶向,我们首先测定每个位点在hek293细胞中的dsdna切割。使用侧接靶位点的pcr引物,我们使用高保真聚合酶(neb,phusion)来扩增基因组序列,其然后通过t7ei测定测试切割活性;我们照常规将靶向效率鉴定在30-75%之间。在这些位点已确认其诱导切割的能力后,将它们克隆到我们构建的sacas9切口酶靶向质粒内,所述质粒含有用于表达四个grna的四个h1启动子盒。随后,在hek293细胞中执行通过pcr的缺失分析,以评价靶向构建体的效率。从我们的生物信息学预测的脱靶基因座就假诱变进行评价。
3.关于中靶/脱靶诱变的高流通量测序。如上所述,我们还对于我们的sacas9靶向实验执行位点特异性深度测序分析。鉴于我们正在使用切口酶方法,我们不预期检测到脱靶诱变,然而,我们已测定替代sacas9靶向位点,其是用于生成隐藏外显子的内含子缺失的潜在替代方案。
4.aav病毒生产。这将如上所述执行。
lca10干细胞系的生成
4背景和原理。虽然小鼠可以充当关于视网膜变性的**模型,但由cep290中的突变引起的rd16小鼠不是用于测试crispr/aav治疗剂的合适模型。与人点突变不同,小鼠变性表型由大的(897bp)纯合框内缺失引起36。因此,为了更好地反映人疾病,我们目前正在使用两种方法来生成lca10细胞:1)将ivs26c.2991+1655a>g突变改造到h7人胚胎干细胞系内,以及2)衍生自lca10患者成纤维细胞的同基因ips细胞系。
实验设计与方法。
1.基因编辑的hesc系。我们已使用crispr-cas9成功地改造点突变,使用~150个碱基寡核苷酸供体(侧翼同源性的~75个碱基)。编码cas9、上述grna序列和供体寡核苷酸的质粒将通过电穿孔引入(ranganathan,v.等人(2014)naturecommunications5,4516)。转染效率通过电穿孔后24小时的荧光进行监测,并且通过t7ei测定评价大量基因靶向。更后,重组克隆通过pcr筛选所需插入,然后通过桑格测序加以验证。分离携带a>g突变的多重纯合和杂合系,并且使用上述生物信息学评价脱靶诱变。
2.患者衍生的ipsc系。与johnshopkinswilmerretinaldegenerationclinic合作,我们目前正在寻找携带cep290突变的患者成纤维细胞。一旦鉴定,就根据由jhmi-sominstitutionalreviewboard的现有irb批准的协议,在知情同意书后收集皮肤活检样品。lca10患者衍生的ipsc将使用已建立的方案(galluzzi,l.等人(2012)celldeathanddifferentiation19,107-120;yu,j.等人(2007)science318,1917-1920;takahashi,k.等人(2006)cell126,663-676;ludwig,t.e.等人(2006)natbiotechnol24,185-187)制备。使用上述技术,我们将平行生成突变校正的同基因细胞系,并且预测的脱靶基因座将进行桑格测序以验证外来突变的不存在。
生物伦理学。我们将使用人胚胎干细胞系h7(wicell);这些细胞系用于所提出的实验的使用由jhuiscro委员会(申请iscro00000023)批准。另外,我们将在jhu机构审查委员会(irb#na_00047271)的主持下,在知情同意书下使用在我们实验室生成的hipsc。尽管不视为es细胞系,但我们仍遵循由jhuembryonicstemcellresearchoversightcommittee规定的所有政策和法规。
3.基因编辑的hesc系的体外编辑和/或患者衍生的ipsc系的编辑。为了彻底评价上文开发的各种先导病毒载体,将测试来自患者衍生的ipsc细胞系或基因编辑的hesc细胞系的细胞;靶向lca10突变的构建体直接处理wtspcas9、espcas9或spcas9-hf,以及使用sacas9切口酶的构建体处理四个grna(图56)。首先,中靶切割效率通过t7ei测定进行测定。对于高分辨率分析,miseq用于定量中靶和脱靶诱变。因为spcas9靶向依赖易错nhej来消除用于剪接的关键核苷酸,所以该分析提供治疗潜力的良好指示。除切割效率和脱靶诱变之外,qrtpcr还用于定量突变型和野生型等位基因两者的cep290表达水平。这提供在验证我们的构建体中的**步。使用我们在实验室中的现有能力,可以使用我们的高内容物机器执行定量纤毛发生测定(kim,j.等人(2010)nature464,1048-1051),以评价我们的治疗构建体的潜力。另外,我们已开发了用于将干细胞分化成光感受器的方案,其允许我们评价相关细胞类型中的表型拯救。鉴于缺乏用于评价关于lca10的基因组编辑的良好动物模型,我们认为这些分析更紧密地接近我们病毒构建体的真正治疗潜力。
实施例6
aav5通过视网膜下注射递送至p0.5小鼠。在28天的任何14天后,收获视网膜且执行t7endoi测定。(注意:aav5靶向视杆光感受器,并且测定对总视网膜执行)。
对于t7endoi分析,通过在quickextract溶液(epicentre,madison,wi)中重悬浮细胞,在65℃下温育20分钟,然后在98℃下温育20分钟,来提取基因组dna。提取溶液直接使用或使用dnacleanandconcentrator(zymoresearch,irvine,ca)清洁,并且通过nanodrop(thermo30fisherscientific)定量。使用phusiondna聚合酶(newenglandbiolabs),从~100ng基因组dna扩增crispr靶位点周围的基因组区域。合并多重独立的pcr反应物,并且使用dnacleanandconcentrator(zymoresearch,irvine,ca)纯化。使在10mmtris-hcl、50mmnacl、10mmmgcl2和100μg/mlbsa中含有150ngpcr产物的25μl体积变性,并且缓慢再退火以允许形成异源双链体:95℃10分钟,95℃至85℃以-1.0℃/秒渐变,85℃1秒,85℃至75℃以-1.0℃/秒渐变,75℃1秒,75℃至65℃以-1.0℃/秒渐变5,65℃1秒,65℃至55℃以-1.0℃/秒渐变,55℃1秒,55℃至45℃以-1.0℃/秒渐变,45℃1秒,45℃至35℃以-1.0℃/秒渐变,35℃1秒,35℃至25℃以-1.0℃/秒渐变,然后保持在4℃下。向每个反应中加入1μlt7endoi(newenglandbiolabs),在37℃下温育30分钟,然后立即置于冰上。对于凝胶分析,将3μl反应物与3μl2x上样染料(newenglandbiolabs)混合,上样到6%tbe-page凝胶上,并且在显现之前用sybrgold(1:10,000)染色~15分钟。凝胶在gellogic200imagingsystem(kodak,15rochester,ny)上显现,并且使用imagejv.1.46定量。使用二项式导出的方程计算nhej频率:
%基因修饰=100*(1-(sqrt(1-((a+b)/(a+b+c)))))
其中“a”和“b”的值等于在本底扣除后的切割片段的积分面积,并且“c”等于在本底扣除后未切割的pcr产物的积分面积。
1.levkovitch-verbinh等人iovsorg44,3388–3393(2003).
2.howellgr等人jcellbiol179,1523–1537(2007).
3.adalbertr等人science(2012),doi:10.1126/science.1223899.
4.yangj等人cell160,161–176(2015).
5.welsbieds等人procnatacadsciusa110,4045–4050(2013).
6.watkinsta等人procnatacadsciusa110,4039–4044(2013).
7.barresba等人neuron1,791–803(1988).
8.millerbr等人natneurosci12,387–389(2009).
9.petrs-silvah等人molther19,293–301(2011).
10.petrs-silvah等人molther17,463–471(2009).
11.sunh等人molvis17,864–875(2011).
12.ribasvt等人neuroscience180,64–74(2011).
13.davismi等人natbiotechnol29,1046–1051(2011).
14.karamanmw等人natbiotechnol26,127–132(2008).
15.bounoutasa等人procnatacadsciusa108,3982–3987(2011).
16.takiharay等人investophthalmolvissci52,3039–3045(2011).
17.jacksonal等人rna12,1179–1187(2006).
18.jacksonal等人natbiotechnol21,635–637(2003).
19.marinesjournalofbiomolecularscreening17,370(2011).
20.hannusm等人nucleicacidsres42,8049–8061(2014).
21.chens等人cell159,440(2014).
22.buehlere等人scirep2,428(2012).
23.yoshidak等人investophthalmolvissci43,1631–1635(2002).
24.bhoumika等人procnatacadsciusa101,4222–4227(2004).
25.jiangy等人jbiolchem288,18429–18438(2013).
26.linl等人devdyn240,52–64(2010).
27.okamotos等人procnatacadsciusa97,7561–7566(2000).
28.ornatskyoi等人journalofbiologicalchemistry272,33271–33278(1997).
29.shalizia等人science311,1012–1017(2006).
30.akhtarmw等人plosone7,e34863(2012).
31.xiongx等人jcellbiol191,211–223(2010).
32.fernandeska等人neurobioldis69,108–116(2014).
33.delimas等人procnatacadsciusa109,9149–9154(2012).
34.smithpd等人neuron64,617–623(2009).
35.sunf等人nature480,372–375(2011).
36.babettoe等人cellrep3,1422–1429(2013).
37.huntwork-rodriguezs等人jcellbiol202,747–763(2013).
38.culicansm等人molcellneurosci41,304–312(2009).
39.vobq等人visneurosci28,175–181(2011).
40.yand等人neuron76,534–548(2012).
41.kampmannm等人procnatacadsciusa112,e3384–91(2015).
42.gonzálezf等人cellstemcell15,215–226(2014).
43.wongn等人genomebiol16,218(2015).
44.doenchjg等人natbiotechnol32,1262–1267(2014).
45.yangl等人natcommun5,5507(2014).
46.varshneygk等人genomeres25,1030–1042(2015).
47.wangth等人jbiolchem273,4928–4936(1998).
48.huj等人biochimicaetbiophysicaacta(bba)-proteinsandproteomics1844,224–231(2014).
49.fiedlerd等人cell136,952–963(2009).
50.wucc等人cellcycle14,1207(2015).
51.swiechl等人natbiotechnol33,102–106(2015).
52.ranfa等人nature520,186–191(2015).
53.ranganathanv等人natcommun5,4516(2014).
54.baerm等人nucleicacidsres18,97–103(1990).
55.myslinskienucleicacidsres29,2502(2001).
56.koilkondar等人molvis15,2796(2009).
57.bemelmansap等人plosone8,e61618(2013).
58.byrnelc等人molther23,290(2014).
59.hiraisi等人jneurosci31,6468–6480(2011).
60.beif等人cell164,219–232(2016).
61.ramakrishnas等人natcommun5,3378(2014).
62.tsaisq等人natbiotechnol33,187–197(2015).
63.parsonsbd等人plosone4,e8471(2009).
64.slaymakerim等人351,84–88(2015).
65.kleinstiverbp等人nature(2016).
66.fuy等人natbiotechnol32,279–284(2014).
67.ranfa等人cell154,1380–1389(2013).
68.potthoffmjdevleopment134,4131–4140(2007).
69.okamotosi等人procnatacadsciusa99,3974–3979(2002).
70.gongx等人neuron38,33–46(2003).
71.flavellsw等人science311,1008–1012(2006).
72.thomassn等人analchem87,10830–10838(2015).
73.aiyetanp等人bmcbioinformatics16,411(2015)。
参考文献
本说明书中提及的所有出版物、**申请、**和其他参考文献指示了本文公开主题所属领域的技术人员的水平。所有出版物、**申请、**和其他参考文献都通过引用并入本文,其程度如同每个个别出版物、**申请、**和其他参考文献明确地且个别地指出通过引用并入。应理解,尽管本文提及了许多**申请、**和其他参考文献,但此类参考文献并不构成这些文献中任一形成本领域公知常识的部分的承认。
原文链接:优钢网 » 用于治疗视网膜变性的基于CRISPR/CAS9的组合物和方法与流程




发表评论